Role of the oxalate transporter SLC26A6 in maintaining kidney health
Student

Prinicipal Investigator

PD Dr. Martin Reichel
Dr. Harald Stachelscheid
Scientific interest within the context of the graduate college:
Our laboratory focuses on the mechanisms involved in maintaining oxalate homeostasis. Oxalate is a component of various foods and is absorbed via the intestine. High urinary oxalate concentrations lead to kidney stones, the second most common kidney disease after hypertension. Furthermore, we have shown that elevated blood oxalate concentrations are associated with cardiovascular disease.1 We are working translationally and recently demonstrated that oxalate uptake in the intestine can be reduced via an enzyme isolated from bacteria in patients.2
Our research group has cloned the first oxalate transporter (SLC26A6).3 SLC26A6 is expressed in different organs. The transporter is located on the apical side of epithelia and actively secretes oxalate into the intestinal lumen4 and urine.5,6 Via this transport process, the oxalate concentration in the body is kept low. If the transporter is missing, there is an increased uptake of oxalate from the intestine, and consequently the formation of kidney stones and progressive kidney damage.7
The subject of our current scientific work is the role of oxalate in the kidney and specifically its influence on the progression of renal diseases. For this purpose, we are working with pluripotent stem cells from which kidney organoids are derived as models.
Specifically, the question of our proposed thesis project is to what extent oxalate has toxic effect(s) on kidney cells. Here we have concrete preliminary data that an accumulation of oxalate has an intracellular toxic effect and that deletion of oxalate transporter SLC26A6 promotes cell death.
Project description:
WP 1: Characterization of renal organoids from pluripotent stem cells. SLC26A6 deficient human pluripotent stem cells have been generated using CRISP/Cas technology in collaboration with BIH Stem Cell Core. As a first step, you will 1) grow kidney organoids and 2) characterize the organoids using kidney cell markers.
WP 2: Examine the effect of oxalate accumulation using kidney organoids. You will be exposing human kidney organoids to oxalate in the presence and absence of the oxalate transporter SLC26A6. You will be examining the mechanism of cell death induced by oxalate by the 1) release of LDH, 2) changes in membrane integrity and 3) the activation of Casp-3 (by western blotting, immunohistochemistry of cleaved Casp-3, and measurement of cleavage activity using fluorescent substrate).
References
- Pfau A, Ermer T, Coca SG, […], Aronson PS, Drechsler C, Knauf F. High Oxalate Concentrations Correlate with Increased Risk for Sudden Cardiac Death in Dialysis Patients. J Am Soc Nephrol. 2021; 32:2375-2385, doi: 10.1681/ASN.2020121793.
- Lieske JC, Lingeman JE, Ferraro PM, […], Tosone C, Kausz AT, Knauf F. Randomized Placebo-Controlled Trial of Reloxaliase in Enteric Hyperoxaluria. N Engl J Med Evid. 2022. doi: 10.1056/EVIDoa2100053.
- Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc Natl Acad Sci USA. 2001; 98:9425-9430, doi: 10.1073/pnas.141241098.
- Neumeier LI, Thomson RB, Reichel M, Eckardt KU, Aronson PS, Knauf F. Enteric Oxalate Secretion Mediated by Slc26a6 Defends against Hyperoxalemia in Murine Models of Chronic Kidney Disease. J Am Soc Nephrol. 2020; 31:1987-1995. doi: 10.1681/ASN.2020010105.
- Knauf F, Ko N, Jiang Z, […], van Itallie CM, Anderson JM, Aronson PS.Net intestinal transport of oxalate reflects passive absorption and SLC26A6-mediated secretion. J Am Soc Nephrol. 2011; 22:2247-2255. doi: 10.1681/ASN.2011040433.
- Knauf F, Thomson RB, Heneghan JF, […], Egan ME, Alper SL, Aronson PS. Loss of Cystic Fibrosis Transmembrane Regulator Impairs Intestinal Oxalate Secretion. J Am Soc Nephrol. 2017; 28:242-249. doi: 10.1681/ASN.2016030279.
- Jiang Z, Asplin JR, Evan AP, […], Nottoli TP, Binder HJ, Aronson PS. Calcium oxalate urolithiasis in mice lacking anion transporter Slc26a6. Nat Genet. 2006; 38:474-478. doi: 10.1038/ng1762.
Innate lymphoid cells, IL-22 and liver regeneration


Scientific interest within the context of the graduate college:
We study development and function of the innate immune system, in particular of innate lymphoid cells (ILC). A current focus is to obtain a molecular understanding of how the innate immune system, by integrating environmental signals (such as those derived from nutrients, microbiota, circadian rhythm) contributes to tissue physiology. Recent studies have revealed ever more intriguing relationships between innate immune system components and basic developmental and biologic processes that are likely to reveal unsuspected pathways by which the immune system might be plumbed to improve health and healthspan. These lines of research have suggested new functions of the immune system for processes such as tissue homeostasis, morphogenesis, metabolism, regeneration and growth. Our research is developing by crossing boundaries of disciplines (immunology, microbiology, developmental biology, stem cell biology, nutrition sciences, tumor biology, regenerative medicine etc.) and is, by nature, highly interdisciplinary.
Project description:
Innate lymphoid cells (ILC) are tissue-resident innate lymphocytes that are involved in immunity to infections but are also deeply integrated in the regulation of tissue function. Based on our preliminary and on published data (Gronke, Nature 2019; Guendel, Immunity 2020; Diefenbach, Immunity 2020), we hypothesize that ILC regulates the function of non-hematopoietic cells to adapt organ function. In this project, we are exploring the role of ILC3 and of IL-22 in liver regeneration. Our preliminary data show that liver regeneration is dependent on IL-22. We have already obtained a high-resolution scRNAseq atlas of the regenerating liver of wildtype mice and of IL-22-deficient mice that reveal molecular network of IL-22-dependent regeneration. Three key questions will be addressed: (1) How is IL-22 production regulated during liver regeneration? Preliminary data reveal a neuron-ILC-hepatocyte axis that controls hepatocyte differentiation and renewal. (2) Which key regenerative pathways in hepatocytes are controlled by IL-22? Key data indicate that IL-22 signaling promotes a Wnt-driven regenerative program. (3) Can IL-22 be used to promote liver regeneration? We use mouse genetics combined with CRISPR/Cas9-driven lineage tracing/barcoding and high-dimensional single-cell genomics.
References
- Witkowski M, Tizian C, Ferreira-Gomes M, […], Radbruch A, Mashreghi MF, Diefenbach A. Untimely TGFβ responses in severe COVID-19 limit antiviral function of NK cells. Nature. 2021; 600:295-301. doi: 10.1038/s41586-021-04142-6.
- Schaupp L, Muth S, Rogell L, […], Schild H, Diefenbach A1,*, Probst HC*. Microbiota-induced tonic type I interferons instruct a poised basal state of dendritic cells. Cell. 2020; 181:1080-1096. doi: 10.1016/j.cell.2020.04.022. 1lead senior author; *equal contribution.
- Guendel F, Kofoed-Branzk M, Gronke K, […], Mashreghi MF, Kruglov AA, Diefenbach A. Group 3 innate lymphoid cells program a distinct subset of IL-22BP-producing dendritic cells demarcating solitary intestinal lymphoid tissues. Immunity. 2020; 53:1015-1032. doi: 10.1016/j.immuni.2020.10.012.
- Gronke K, Hernández PP, Zimmermann J, […], Glatt H, Triantafyllopoulou A, Diefenbach A. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. 2019; 566:249-253. doi: 10.1038/s41586-019-0899-7.
- Hernandez P, Mahlakoiv T, Yang I, […], Suerbaum S, Staeheli P, Diefenbach A. Interferon-λ and interleukin-22 cooperate for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol. 2015; 16:698-707. doi: 10.1038/ni.3180.
The role of DNA damage response signaling in chronic kidney disease


Scientific interest within the context of the graduate college:
Our research group studies the underlying mechanisms of chronic kidney disease (CKD). Acute and chronic kidney injury has been increasingly recognized as a global public health concern, associated with high morbidity, and mortality. Acute kidney injury is frequent, occurring in 21% of hospital admissions and leads to CKD regardless of the cause. CKD encompasses a group of heterogeneous disorders affecting renal structure and function with a prevalence of 10-15% worldwide1. During the past decades, research into kidney disease has largely focused on identifying causative insults and disease modifiers of renal injury. However, sufficient clarification about the underlying pathophysiological mechanism has not provided yet.
We hypothesize that the ability of the kidney to respond to different stress conditions contributes significantly to the maintenance of normal renal function and structure, whereas an impaired cellular stress responses promotes renal damage. Here, we focus on the so-called “DNA Damage Response” (DDR)2, which relevance for kidney diseases has been demonstrated in individuals with interstitial nephritis caused by monogenic mutations in genes encoding proteins of the DDR complex3. Interestingly, transgenic mice models with a defective DDR response show an increased susceptibility to environmental nephrotoxins, which leads to renal failure and typical histological features of CKD4. Moreover, recently published data argues that DDR functions are critical in disease progression of rare, inherited juvenile nephropathies as evidenced by accumulated DNA damage, that yields to increase apoptosis coupled with profibrotic responses5,6. This projects aims to investigate if an impaired DNA damage response contributes to the renal pathomechanisms of AKI and CKD.
Project description:
In CKD, regardless of its cause, renal fibrosis is the primary determinant of end-stage kidney disease, with no effective therapy available today. Within in scope of this project it is planned to study the response of kidney tubular cells to DNA damage, which may play a role in the pathophysiology of CKD. Therefore, we employed a human in-vitro model using patient derived renal tubular cells from urine samples, which allows investigating the biological impact of DNA damage response signaling. In a comparative approach, cultured renal tubular cells from individuals with CKD and healthy controls will be used to study the cellular response by applying different molecular techniques. In detail, it is planned to evaluate the activity of the DDR pathway targeting e.g. the accumulation of DNA damage, the cell cycle progression, the rate of apoptosis and profibrotic gene expression profiles. This project provides a new cellular model to study disease mechanisms of acute and chronic kidney injury and may offer new understandings about the pathogenesis of CKD.
References
- Eckardt KU, Coresh J, Devuyst O, […], Köttgen A, Levey AS, Levin A. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet. 2013; 382:158-169. doi: 10.1016/S0140-6736(13)60439-0.
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010; 40:179-204. doi: 10.1016/j.molcel.2010.09.019.
- Zhou W, Otto EA, Cluckey A, […], Levy S, Smorgorzewska A, Hildebrandt F. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. 2012; 44:910-915. doi: 10.1038/ng.2347.
- Airik R, Schueler M, Airik M, […], Mukherjee E, Sims-Lucas S, Hildebrandt F. A FANCD2/FANCI-Associated Nuclease 1-Knockout Model Develops Karyomegalic Interstitial Nephritis. J Am Soc Nephrol. 2016; 27:3552-3559. doi: 10.1681/ASN.2015101108.
- Chaki M, Airik R, Ghosh AK, […], Smogorzewska A, Otto EA, Hildebrand F. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012; 150:533-548. doi: 10.1016/j.cell.2012.06.028.
- Slaats GG, Giles RH. Are renal ciliopathies (replication) stressed out? Trends Cell Biol. 2015; 25:317-319. doi: 10.1016/j.tcb.2015.03.005.
Cellular senescence is a common pattern in disease models of different forms of steatotic liver disease
Student

Prinicipal Investigator

Prof. Dr. Frank Tacke
Scientific interest within the context of the graduate college:
Cellular senescence occurs in the liver in health and disease. Senescence relates to a status of cell cycle arrest, which becomes more prevalent with increasing age and which develops as the consequence of liver disease.1 Due to subsequent changes in cell morphology and functionality senescent cells may prompt disease progression and the development of disease-related complications.1-3 Detection of senescence and deciphering of its underlying mechanisms may help identifying novel targets to develop preventive treatment strategies to halt the development of liver disease related complications such as fibrosis, inflammation, and ultimately cirrhosis.4 Therefore, this project seeks to address the following aspects:
- Targeting (hepatocellular) senescence may be considered as a preventive strategy to maintain health by halting disease progression to irreversible stages early in the beginning of disease development.
- Molecular pathways initiating hepatocellular senescence will be described and analyzed in great detail in order to lay the basis for novel interceptive therapies preventing liver diseases to establish.
- Ideal time points for potential interventions (senolysis) to restore health and to prevent accelerated aging as the consequence of diseases will be explored.
Project description:
Aim: Cellular senescence is a well-known consequence of diseases in general but in-depth characterization of triggers (DNA damage, Telomere shortening, etc.), pathways leading to cell cycle arrest (e.g. p16 dependent pathway, p53 dependent pathway), and subsequent cell-phenotypic changes (e.g. GATA4 driven SASP, Cyp450 expression, metabolic liver zonation) are sparse. Therefore, the main aim of this project is to decipher the characteristics of hepatocytes senescence in different types and stages of liver disease. It will serve as the basis for any type of senolytic intervention, first with respect to the type of senolytic compound [broad spectrum (e.g. BCL2 inhibitor) vs. pathway specific (e.g. p53 interacting proteins)] and second, with respect to the time point of intervention. It will be based on an in-depth analysis of human and rodent tissue samples.
Workplan: Healthy livers from different ages will be included to understand to what extent aging drives cellular senescence in the liver. The following liver diseases are relevant in Europe thus being involved in this project: 1) Paracetamol-induced liver injury 2) Drug-induced liver injury 3) Alcoholic hepatitis 4) Non-alcoholic steatohepatitis 5) Cirrhosis 6) Acute-on-chronic liver failure (ACLF). Human FFPE liver samples with different disease stages from the Pathology department Charité will be analyzed by conventional immunostaining and multiplex immunofluorescence staining with respect to DNA-damage, cell cycle arrest, autophagy, and cell death. Image analysis will be attempted to be on single-cell level including descriptive neighboring analysis. In addition, whilst regenerative mechanisms generally appreciate the proliferative activity of cells and preservation of tissue integrity, restoration of metabolic activity by alteration of liver zonation to maintain organ function is another incremental part of liver regeneration although being frequently disregarded. Therefore, expression of senescence markers will be correlated with alteration of liver zonation and metabolic characterization of hepatocytes. Mechanistic information will be obtained from already performed animal models for the above mentioned diseases as well as conditional knockout mouse lines [AhCreMdm2fl/fl (inducing senescence in hepatocytes); AhCrep53fl/fl (preventing senescence in hepatocytes)] after exposure to toxins such as lipopolysaccharides or ethanol. In addition to immunostaining analyses from paraffin-embedded tissue, frozen tissue will allow generating proteomic (mass spectrometry) and transcriptomic (mRNA sequencing) data. By using laser capture microdissection to extract proteins and RNA from areas of interest spatial information can be added to this analysis. Performing Seahorse from frozen section of liver tissue will generate more data on the metabolic/mitochondrial tissue function and will be correlated with changes of metabolic liver zonation and presence of cellular senescence in different liver diseases. All experimental techniques are well established in our lab and respective animal experiments were performed previously.
Impact: Results from this project will provide highly relevant information regarding the role of cellular senescence in homeostasis and different types of liver disease, will allow generating disease related hypothesis and may serve as the basis for later interventional study (e.g. senolytic therapies) aiming at restoring “liver health” with respect to timing of intervention and choice of senolytic compounds.
References
- Ferreira-Gonzalez S, Rodrigo-Torres D, Gadd VL, Forbes SJ. Cellular Senescence in Liver Disease and Regeneration. Semin Liver Dis. 2021; 41:50-66. doi: 10.1055/s-0040-1722262.
- Kang C, Xu Q, Martin TD, […], Yanker BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015; 349:aaa5612. doi: 10.1126/science.aaa5612.
- Engelmann C,Tacke F. The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. Int J Mol Sci. 2022; 23:652. doi: 10.3390/ijms23020652.
- Baar MP, Brandt RMC, Putavet DA, […], Hoeijmakers JHJ, Campisi J, de Keizer PLJ. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell. 2017; 169:132-147 e116, doi: 10.1016/j.cell.2017.02.031
Learning from outliers – Identifying determinants of kidney survival in non-progressive ADPKD


Scientific interest within the context of the graduate college:
Not Everything Is “Genetic”, but Genes Are Involved in Everything (adapted from Kenneth M. Weiss). Our group is interested in identification and investigation of genetic, clinical, and environmental factors determining onset of chronic kidney disease (CKD) and kidney survival. We make use of next-generation sequencing techniques and deep-phenotyping to identify genetic variants that are predictive for disease progression or convey protection from organ failure. We functionally evaluate identified germline variants in vitro in order to understand underlying molecular mechanisms leading to CKD on the one hand or protecting from kidney failure on the other. By doing so, we aim at defining and targeting molecular switches responsible for health maintenance and disease alleviation.
Project description:
One of six patients undergoing renal transplantation has autosomal-dominant polycystic kidney disease (ADPKD) caused by heterozygous germline mutations in one of two main disease genes, namely PKD1 (encoding polycystin 1, PC1) or PKD2 (encoding polycystin 2, PC2).ADPKDis the commonest genetic disorder leading to CKD including end-stage kidney failure (ESKF).1-3 ESKF from ADPKD commonly occurs between ages 30-80 years. While PKD1-associated disease is generally more severe than PKD2-disease, exemplified by a 20-year difference in mean age at ESKF (55 versus 75 years), current genotype-phenotype correlations are still crude.4,5 For example, some patients with even identical PKD1-mutations vary dramatically in their progression. We demonstrated that additional non-diagnostic hypomorphic PKD1-germline variants, as well as variants in genes involved in proteostasis mechanistically, add to the PKD1-mutational effect.6,7 The underlying mechanisms involve posttranslational modification and endoplasmic reticular (ER)-processing of PC1. As a result, PC1 expression at the cell surface is reduced.8 Conversely, opposite mechanisms may confer renoprotection by increasing PC1 surface expression. We aim to learn from clinical outliers and hypothesize that the latter mechanisms play a role in individuals with ADPKD not depending on dialysis until their 70s and beyond. To address this, we propose the following two specific aims and work packets (WP):
Aim 1/WP1: Identification of genetic determinants associated with non-progression and kidney survival. Exome sequencing in individuals (n=20) with mildest, non-progressive ADPKD-PKD1 (non-ESKF > 70 yrs) and most severe ADPKD-PKD1 (ESKF < 40 yrs) from the Leipzig-Berlin ADPKD-cohort (n=400) via the BIH-sequencing core facility. Consecutive assessment for additional rare and common variants (single variant analysis and variant burden analysis) by use of a predefined CKD/ADPKD-candidate gene panel. Identification of differentially enriched candidate gene sets and single variants, suitable for functional analysis (WP2).
Aim 2/WP2: Validation of genetic determinants associated with non-progression and kidney survival. Overexpression of two most promising gene variants significantly associated with kidney survival and consecutive use of established cellular read-outs on RNA and protein level. Planned analyses include qRT-PCR, Western Blot, and immunofluorescence imaging (e.g. PC1 surface expression).
References
- Lanktree MB, Haghighi A, Guiard E, […], Harris PC, Paterson AD, Pei Y. Prevalence Estimates of Polycystic Kidney and Liver Disease by Population Sequencing. J Am Soc Nephrol. 2018; 29: 2593-2600. doi: 10.1681/ASN.2018050493.
- Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019; 393:919-935. doi: 10.1016/S0140-6736(18)32782-X.
- Schönauer R, Baatz S, Nemitz-Kliemchen M, […], Neuber S, Bergmann C, Halbritter J. Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet Med. 2020; 22:1374-1383. doi: 10.1038/s41436-020-0816-3.
- Su Q, Hu F, Ge X, […], Zhou Q, Mei C, Shi Y. Structure of the human PKD1-PKD2 complex. Science. 2018; 361: eaat9819. doi: 10.1126/science.aat9819.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. New Engl J Med. 2011; 364:1533-1543. doi: 10.1056/NEJMra1010172.
- Irazabal MV, Rangel LJ, Bergstralh EJ, […],BJ, King BF, Torres VE, CRISP Investigators. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015; 26:160-172. doi: 10.1681/ASN.2013101138.
- Zhang Z, Bai H, Blumenfeld J, […], Robinson RD, Kapur S, Rennert H. Detection of PKD1 and PKD2 Somatic Variants in Autosomal Dominant Polycystic Kidney Cyst Epithelial Cells by Whole-Genome Sequencing. J Am Soc Nephrol. 2011; 32:3114-3129. doi: 10.1681/ASN.2021050690.
- Durkie M, Chong J, Valluru MK, Harris P C, Ong A C M. Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease. Genet Med. 2021; 23:689-697. doi: 10.1038/s41436-020-01026-4.
Role of Oncostatin M in acute pulmonary inflammation


Scientific interest within the context of the graduate college:
The mammalian gastrointestinal tract contains the largest number of immune cells and harbors a large and diverse population of commensal bacteria that exist in a symbiotic relationship with the host.1,2 The gut-resident immune cells are separated from our microbial residents by a single layer of intestinal epithelial cells (IEC). The dynamic cross-talk between IEC, the intestinal microbiota, and local immune cells represents a cornerstone of intestinal homeostasis.3,4 The balance between the various immune cell populations and tonic cytokine signals play an important role in determining thresholds of tolerance and immunity in the intestine.
Project description:
We recently highlighted the relevance of Oncostatin M (OSM) in intestinal inflammation.5 OSM is a pleiotropic cytokine belonging to the interleukin 6 (IL-6) family, which influences numerous homoeostatic and pathological processes in various organs, yet its biology remains obscure.6,7 OSM receptor (OSMR) is widely expressed at both tissue (vascular system, heart, lung, adipose tissue, skin, bladder, mammary tissue, adrenal gland, and prostate) and cellular levels (endothelial, smooth muscle, fibroblast, and lung epithelial cells). In contrast, OSM is expressed in multiple hematopoietic cell types including activated monocytes/macrophages, neutrophils, dendritic cells, and T cells. We showed recently that OSMR is widely expressed by stromal and endothelial cells in the intestine; however, the role of OSM in the maintenance of intestinal homeostasis remains unknown. We hypothesize that microbiota-derived local cues induce constitutive OSM expression by gut-resident immune cells to promote intestinal homeostasis by acting on both stromal and endothelial cell compartments. This project will exploit new reporter mouse lines generated in Hegazy lab (OsmriScarlet, OsmZsgreen), gnotobiotic mice, and primary human tissue samples to explore the signals regulating OSM expression in the gut and how OSM functions as a potential tissue rheostat. The project will utilize different cellular and molecular biology techniques, including magnetic cell isolation, flow cytometry, gene expression analysis, RNA sequencing, and histology.
The following specific objectives will be addressed in this project:
- Characterize the influence of intestinal microbiota on the regulation of OSM pathway
- Identify the down-stream sensing pathways regulating OSM-OSMR axis in gut-resident immune cells
- Assess the relevance of the OSM-OSMR pathway in promoting intestinal homeostasis
References
- Belkaid Y, Hand TW. Role of the Microbiota in Immunity and Inflammation. Cell. 2014; 157:121-141. doi: 10.1016/j.cell.2014.03.011.
- Macpherson AJ, Slack E, Geuking MB, McCoy KD. The mucosal firewalls against commensal intestinal microbes. Semin Immunopathol. 2009; 31:145-149. doi: 10.1007/s00281-009-0174-3.
- Hooper LV, Littman DR, Macpherson AJ. Interactions Between the Microbiota and the Immune System. Science 2012; 336:1268-1273. doi: 10.1126/science.1223490.
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014; 14:141-153. doi: 10.1038/nri3608.
- West NR, Hegazy AN, Owens BMJ, […], Keshav S, Travis SPL, Powrie F. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med. 2017; 23:579-589. doi: 10.1038/nm.4307.
- Richards CD. The Enigmatic Cytokine Oncostatin M and Roles in Disease. ISRN Inflamm. 2013; 2013:512103. doi: 10.1155/2013/512103.
- West NR, Owens BMJ, Hegazy AN. The oncostatin M-stromal cell axis in health and disease. Scand J Immunol. 2018; 88(3):e12694. doi: 10.1111/sji.12694.
Characterization of the peripheral immune compartment during fasting in healthy volunteers and MS patients


Scientific interest within the context of the graduate college:
Diet is an important factor for a healthy life. For the most part of human history, the next meal was not a given. Hence, there was a strong selection pressure for adaptation to periods of no or low food consumption during our evolution. Consequently, today’s excessive calorie intake, as it is typical for diets in the western world, results in increasing occurrence of systemic inflammation and widespread diseases. In contrast, calorie restriction has been shown to improve numerous chronic diseases and to prolong the healthy lifespan. In my group, we are re-thinking health in the context of evolutionary adaptation to low food energy intake. Specifically, we focus on the identification of cellular and molecular mechanisms how reduced calorie intake maintains health, prevents and improves inflammatory diseases, and prolongs healthy life.
Project description:
The focus of my group is to understand the cellular and molecular mechanisms how reduced calorie intake regulates homeostasis and function of the immune system. Recently, we have found that fasting drastically reduces the number of circulating pro-inflammatory monocytes in the blood of humans and mice.1 Interestingly, monocytes accumulated in the bone marrow. Our preliminary data suggest that this is due to the inhibition of monocyte egress from the bone marrow as well as to recruitment from the blood circulation. This phenomenon can be observed not only for monocytes but also for specific other immune cell populations such as naïve B cells and memory T cells.2,3 Intriguingly, memory T cells that have been re-located to the bone marrow display functional modulation and improvement upon egress and re-circulation.2 Hence, our research question is: Are monocytes functionally modified in the bone marrow during fasting? To answer this question, we will establish a monocyte adoptive transfer model in mice and apply next generation sequencing to monocytes in the periphery and the bone marrow. Thus, this project offers the opportunity to the student to develop bench working skills as well as getting familiar with the computational analysis of large transcriptomic datasets.
The aim of the study is to analyze the transcriptional profile of monocytes that returned to the bone marrow from the periphery during fasting.
WP1: Establishing an adoptive monocyte transfer model into fed and fasted mice. It is crucial to the study that monocytes returning to the bone marrow during fasting can be clearly identified. Therefore, we will isolate Ly-6C+ monocytes from the spleen of CD45.1+ mice using the Miltenyi monocyte isolation kit (Fig. 1). Isolated monocytes will be transferred into CD45.2+ mice that will be fed or fasted for 16 hours via intravenous injection. After 4 hours, bone marrow, blood, spleen, and additional organs will be harvested and analyzed for transferred CD45.1+ monocytes using multicolor flow cytometry. Hence, CD45.1/CD45.2 discrimination will enable us to identify and quantify monocytes that are recruited from the blood to the bone marrow. We already obtained the approval of the responsible state office for the animal experiments.
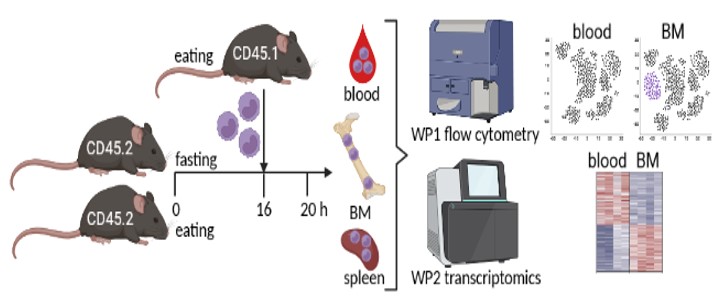
BM = bone marrow.
WP2: Monocyte transcriptomics and computational analysis. In WP2 we will transfer splenic CD45.1+ monocytes into fed and fasted mice as described in WP1 for transcriptional analysis. We will analyze CD45.1+ monocytes that were recruited to the bone marrow, as well as CD45.1+ monocytes that remained in the blood circulation or homed to the spleen. The comparison between monocytes that returned to the bone marrow in fed vs. fasted mice will identify specific fasting-induced modifications. We will also include a monocyte sample from the spleen of a fed mouse for analysis of the pre-transfer state. We will enrich CD45.1+ monocytes from the respective organs using the Miltenyi monocyte isolation kit and subsequently use flow cytometry to sort to high purity for sequencing. Because we do not expect to yield high cell numbers, we will use ultra-low input sequencing approaches and multiplex samples to save resources. The computational analysis will include among others: differentially expressed genes between bone marrow and peripheral monocytes during fasting, gene ontology, KEGG and pathway analysis, upstream regulators of transcriptional changes, prediction of modified cellular functions, analysis of cellular metabolic and transcriptional networks as well as intercellular communication using Ingenuity Pathway analysis and R tools as we have done before.1
In summary, we will investigate functional modifications of monocytes in the bone marrow during fasting using a monocyte transfer model in combination with transcriptional analysis. The results could indicate how fasting reduces the pro-inflammatory potential of monocytes and, thereby, prevents inflammatory disease and prolongs healthy life.
References
- Jordan S, Tung N, Casanova-Acebes M, […], Berres ML, Gallagher EJ, Merad M. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell. 2019; 178:1102-1114 e1117. doi: 10.1016/j.cell.2019.07.050.
- Collins, N, Han SJ, Enamorado M, […], McGavern DB, Schwartzberg PL, Belkaid Y. The Bone Marrow Protects and Optimizes Immunological Memory during Dietary Restriction. Cell. 2019; 178:1088-1101 e1015. doi: 10.1016/j.cell.2019.07.049.
- Nagai M, Noguchi R, Takahashi D, […] Takubo K, Dohi T, Hase K. Fasting-Refeeding Impacts Immune Cell Dynamics and Mucosal Immune Responses. Cell. 2019; 178:1072-1087 e1014. doi: 10.1016/j.cell.2019.07.047.
The role of the epithelial transcription factor NF-κB in homeostasis, development and regeneration of chronic inflammatory bowel diseases


Scientific interest within the context of the graduate college:
Our group “Signal Transduction in Health and Disease” (Department of Gastroenterology and Hepatology, Charité Virchow) aims to understand the molecular mechanisms involved in tissue homeostasis, inflammation, and resolution of inflammation. Our main focus is on the transcription factor NF-κB and its role in the intestinal epithelium. Recent studies showed that crosstalk between epithelium and immune cells changes in health and disease, and that is in part due to altered functions of NF-κB. Our group aims to decipher that crosstalk and identify the changes. Our research is highly interdisciplinary and spans different fields including biochemistry, immunology, stem cell biology, and cancer biology.
Project description:
NF-κB is deemed the master regulator of pro-inflammatory response and its activation directly correlates with severity of inflammation in Inflammatory Bowel Disease (IBD).1 Although in immune cells, NF-κB plays largely pro-inflammatory role, in intestinal epithelium its function is poorly understood. Many IBD therapies rely on suppression of NF-κB. These therapies however inhibit NF-κB across different tissues and cell types, including healthy ones. This can lead to many adverse effects, including susceptibility to infection and carcinogenesis. We have recently shown that a single stress stimulus sequentially activates functionally distinct transcriptomes of NF-κB: anti-apoptotic and pro-inflammatory.2-3 We also showed that in homeostasis, NF-κB directs differentiation of stem cells.4 To identify therapies that specifically target only the pro-inflammatory NF-κB, it is critical to decipher what roles NF-κB plays in health, in acute inflammation, and finally in mucosal healing.
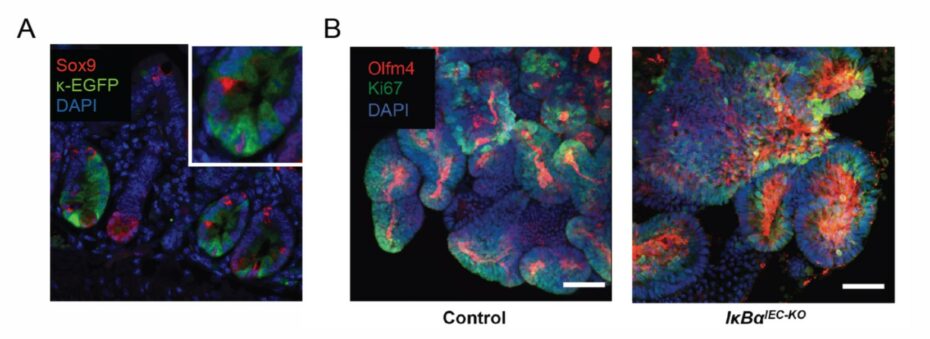
Based on our recently published and unpublished data we propose that acute, immediate activation of NF-κB in intestinal epithelium is necessary for resolution and for regaining homeostasis. To address this hypothesis we will use single-cell datasets, intestinal organoids, and transgenic mouse models. We have recently established transgenic mice that provide a specific readout of NF-kB activity as well as mice with either specific suppression or constitutive activation of NF-kB in intestinal epithelium.2-4 These tools could enable us, for the first time, to determine where and how NF-κB can be targeted to both prevent chronic inflammation or to promote resolution.
Our key aims are:
1) To identify which cells activate NF-kB under homeostasis versus in inflammation, and to determine which transcriptional programs NF-kB initiates in these cells.
2) To determine how repression or constitutive activation of NF-kB affects intestinal homeostasis, acute inflammation, and resolution.
3) Identify which singling pathways can be targeted to safeguard homeostasis or promote resolution of inflammation.
References
- Atreya I, Atreya R, Neurath MF. NF-kappaB in inflammatory bowel disease. J Intern Med. 2008; 263:591-596. doi: 10.1111/j.1365-2796.2008.01953.x.
- Kolesnichenko M, Mikuda N, Höpken UE, […], Schmidt-Ullrich R, Schmitt CA, Scheidereit C. Transcriptional repression of NFKBIA triggers constitutive IKK- and proteasome-independent p65/RelA activation in senescence. EMBO J. 2021; 40:e104296. doi: 10.15252/embj.2019104296.
- Mikuda N, Schmidt-Ullrich R, Kärgel E, […], Scheidereit C, Kühl AA, Kolesnichenko M. Deficiency in IκBα in the intestinal epithelium leads to spontaneous inflammation and mediates apoptosis in the gut. J Pathol. 2020; 251:160-174. doi: 10.1002/path.5437.
- Brischetto C, Krieger K, Klotz C, […], Heuberger J, Scheidereit C, Schmidt-Ullrich R. NF-κB determines Paneth versus goblet cell fate decision in the small intestine. Development. 2021; 148:dev199683. doi: 10.1242/dev.199683.
Cellular mechanisms underlying the pathogenetic role of the primary cilium in pulmonary arterial hypertension


Scientific interest within the context of the graduate college:
Maintenance or restoration of vascular homeostasis are critical determinants of health throughout life. Of late, the primary cilium has been identified as a key regulator of vascular homeostasis and regeneration. While impairment of primary ciliary structure and signaling can promote vascular disease and vascular remodeling, strategies aiming to preserve or restore ciliary function emerge as novel therapeutic approaches to maintain vascular health.
Project description:
Physiologically, homeostatic signaling in response to biochemical and biomechanical cues within the pulmonary vascular wall warrants the integrity of the vascular structure of the lung and allows for its adaptation to changing requirements throughout life. Dysregulated signaling in or between pulmonary artery endothelial and smooth muscle cells, on the other hand, causes progressive adverse remodeling of the pulmonary vasculature, resulting in increased pulmonary vascular resistance, pulmonary hypertension, and ultimately death due to right heart failure. Understanding the cellular signaling mechanisms that maintain or restore vascular homeostasis in the lung is therefore critical for the prevention or therapy of pulmonary hypertension, and for the preservation of an intact vasculature throughout life up to an old age.
Ongoing research in our group has identified the primary cilium as a novel key regulator of vascular homeostasis in the lung. Unlike the motile cilia found e.g. on the respiratory epithelium or in the Fallopian tube, the primary cilium is a singular organelle (one per each cell) that is found on almost every cell type in the body. The primary cilium extends as a protrusion of the cell membrane into the extracellular space where it functions as a mechano- and chemosensor, while its base acts as a hub for numerous cellular signaling pathways. We have shown that the primary cilium maintains the cells of the pulmonary vasculature in a physiological quiescent state. Loss of the primary cilium, however, causes pathological proliferation and migration of both cell types. Importantly, we have identified such loss of the primary cilium as a hallmark of pulmonary blood vessels in patients with pulmonary hypertension. These findings fuel the intriguing hypothesis that strategies aiming to preserve or restore primary cilium structure and function may present a novel therapeutic approach to maintain lung vascular health and to reverse maladaptive vascular remodeling. In order to test this hypothesis, the present project will address the following questions:
Question 1: Which mechanisms regulate the integrity of the primary cilium in lung vascular endothelial and smooth muscle cells? Based on previous work by us and others, we will focus here specifically on signaling axes via mammalian target of rapamycin (mTOR) and aurora kinase A. Specifically, we will test whether pharmacological or genetic modulation of these pathways can maintain primary ciliary integrity in preclinical models of pulmonary hypertension.
Question 2: Can strategies aimed to restore ciliary integrity preserve lung vascular homeostasis? We have recently patented a novel intervention for rescue of the primary cilium in pulmonary hypertension. We will test whether this strategy, as well as interventions identified in Aim 1 can restore vascular homeostasis in preclinical models of pulmonary hypertension both in vitro and in vivo.
References
- Fan Y, Gu X, Zhang J, […], Solymosi P, Kwapiszewska G, Kuebler WM. TWIST1 drives smooth muscle cell proliferation in pulmonary hypertension via loss of GATA-6 and BMPR2. Am J Respir Crit Care Med. 2020; 202: 1283-1296. doi: 10.1164/rccm.201909-1884OC.
- Dummer A, Rol N, Szulcek R, […], DeRuiter MC, Goumans MJ, Hierck BP. Endothelial dysfunction in pulmonary arterial hypertension: loss of cilia length regulation upon cytokine stimulation. Pulm Circ. 2018; 8: 2045894018764629.doi: 10.1177/2045894018764629.
The role of Muc5b in lung homeostasis and onset and progression of interstitial lung disease


Scientific interest within the context of the graduate college:
Mucociliary clearance is the primary innate defense mechanism of the lung and crucial to maintain lung homeostasis and health. Mucociliary clearance relies on motile cilia on the surface of epithelial cells and a protective mucus gel layer entrapping particles and pathogens to be cleared from the lungs. Recent evidence suggests that the secreted mucin MUC5B that is crucial for the formation of the mucus gel and proper mucociliary clearance is also implicated in the pathogenesis of interstitial lung disease (ILD).1 ILD can affect children and adults and is characterized by interstitial inflammation, rapid progression of pulmonary fibrosis and subsequent disruption of the alveolar gas exchange ultimately leading to respiratory failure. The understanding of the pathogenesis remains limited and ILD is usually diagnosed in advanced stages when irreversible lung damage has already occurred. Further, only limited therapeutic options are available, which so far, cannot prevent progression of pulmonary fibrosis. By conditional deletion of Nedd4-2 (Nedd4-2-/-) in lung epithelial cells of mice, we recently generated the first mouse model that develops spontaneous pulmonary fibrosis sharing key features with ILD patients allowing us to study lung homeostasis at baseline and early dysregulation leading to the development of interstitial lung disease.2,3
Project description:
The aim of this translational research project is to investigate the role of Muc5b in the onset and progression of interstitial lung disease in conditional Nedd4-2-/- mice. For this purpose, double knockout mice deficient for Nedd4‑2 and Muc5b will be generated and their lung phenotype comprehensively studied at different stages of disease progression to identify early changes in lung homeostasis and pathways that promote disease progression and may serve as therapeutic targets. Clinical parameters such as inflammation markers, histology, lung function, as well as mucus properties will be investigated in order to gain insight into the relationship between altered mucus properties and disease pathogenesis. Further, our findings will be validated in lung biopsies and serum samples from patients with ILD. In addition to basic molecular biology laboratory work, this experimental MD thesis also applies state-of-the-art mouse lung function, mucus rheological measurements, and high content microscopy.
References
- Evans CM, Fingerlin TE, Schwarz MI, […], Warg L, Yang IV, Schwartz DA. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol Rev. 2016; 96:1567-1591. doi: 10.1152/physrev.00004.2016.
- Duerr J, Leitz DHW, Szczygiel M, […], Beers MF, Klingmüller U, Mall MA. Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat Commun. 2020; 11:2012. doi: 10.1038/s41467-020-15743-6.
- Leitz DHW., Duerr J, Mulugeta S, […], Dalpke AH, Beers MF, Mall MA. Congenital Deletion of Nedd4-2 in Lung Epithelial Cells Causes Progressive Alveolitis and Pulmonary Fibrosis in Neonatal Mice. Int J Mol Sci. 2021; 22:6146. doi: 10.3390/ijms22116146.
How does NK cell memory develop in humans?


Scientific interest within the context of the graduate college:
The innate immune system matures early on along with tissue development. In particular, innate lymphoid cells (ILCs, including Natural Killer (NK) cells, already colonize tissues during fetal life and acquire their effector functions already pre-natally. However, exposure to different environmental signals, including microbial infection, can still remodel ILCs and leave permanent epigenetic marks. Our group focuses on delineating transcriptionally and epigenetically the immune functional programs pre-wired during development as well as those emerged and imprinted after exposure to environmental stimuli, in particular persistent virus, thereby setting the threshold of our immune fitness and tolerance to tissue damage. Our aim is to understand the key homeostatic checkpoints, which, once altered, can initiate inflammatory circuits and lead to disease.
Project description:
Aim 1: Analysis of ex vivo dynamics of NK cell clonality and epigenetic remodeling in response to HCMV. In this part of the project, we aim to understand how NK cell clonal expansions are generated and maintained. To this aim, we will perform the following experiments:
A) Lineage tracing over time of human NKG2C+ and NKG2C– NK cells derived from peripheral blood of HCMV+ and HCMV– healthy individuals by performing scATACseq/scRNAseq and analysis of mitochondrial mutations (DOGMAseq).5
B) Lineage tracing over time of human NKG2C+ and NKG2C– donor NK cells in patients undergoing hematopoietic stem cell transplantation and HCMV reactivation (established cooperation with Hematology Charité, Prof L. Büllinger). NK cells will be isolated from peripheral blood of patients between day 10 and day 30 after transplantation, during HCMV reactivation (typically around day 30), and at later time points (day 60-120). DOGMAseq will be performed as in 1A. Moreover, clinical and laboratory parameters along with sequencing of HCMV-relevant proteins will be assessed.
Aim 2: Analysis of in vitro dynamics of NK cell clonality and epigenetic remodeling in response to HCMV-derived signals. In this part of the project, we aim to understand the signals driving NK cell clonal expansion. To this aim, we will mimic NK cell clonal expansion in vitro by culturing human NKG2C+ “naive” NK cells derived from peripheral blood of HCMV– healthy individuals in the presence of signals mimicking HCMV stimulation (NKG2C peptide ligands, CD2 costimulation, and pro-inflammatory cytokines). Proliferating NK cells will be sorted at different time points and DOGMA-seq will be performed.
References
- Luetke-Eversloh M, Hammer Q, Durek P, […], Chang HD, Dong J, Romagnani C. Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PLoS Pathog. 2014; 10:e1004441. doi: 10.1371/journal.ppat.1004441.
- Hammer Q, Rückert T, Romagnani C. Natural killer cell specificity for viral infections. Nat Immunol. 2018; 19:800-808. doi: 10.1038/s41590-018-0163-6.
- Hammer Q, Rückert T, Borst EM, […], Mashreghi MF, Messerle M, Romagnani C. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat Immunol. 2018; 19:453-463. doi: 10.1038/s41590-018-0082-6.
- Ludwig LS, Lareau CA, Ulirsch JC, […], Buenrostro JD, Regev A, Sankaran VG. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell. 2019; 176:1325-1339.e22. doi: 10.1016/j.cell.2019.01.022.
- Mimitou EP, Lareau CA, Chen KY, Sankaran VG, Regev A, Smibert P. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat Biotechnol. 2021; 39:1246-1258. doi: 10.1038/s41587-021-00927-2.
Investigating the impact of nano- and microplastics on hypertensive disease


Scientific interest within the context of the graduate college:
Environmental factors such as nutrition but also pollution can significantly influence the health-to-disease transition. The contact of the environment with the human organism, especially at interfaces such as the intestine and the immune system, is of crucial importance. Often, several harmful environmental influences come together to trigger a disease or promote its development. Through our project, we aim to create a better understanding of environmental factors that promote the development of cardiovascular diseases via immunological mechanisms.
Project description:
Plastic pollution is a major challenge in our today’s society. Micro- and nanoplastic particles have been found in various foods1 and recently the uptake of the particles into our organism has been proven by the discovery of plastic particles in human blood.2 Microplastics have been shown to alter the microbiome and induce inflammation.3-5 Current research thereby focuses mainly on the impact of plastic in healthy organisms. However, a large amount of our population suffers from diseases such as hypertension. In this project, we aim to investigate the impact of Micro- and nanoplastics in the setting of hypertension. The microbiome-immune axis has been shown to be of high relevance in hypertensive-related organ damage.6,7 We aim to explore the impact of plastic particles on all parts of this axis – the microbiome, the immune system, and organ damage by using a murine hypertension model. The project will help to enlighten the risks of plastic pollution for human health.
References
- Paul MB, Stock V, Cara-Carmona J, […], Braeuning A, Sieg, H, Böhmert, L. Micro- and nanoplastics – current state of knowledge with the focus on oral uptake and toxicity. Nanoscale Adv. 2020; 2:4350-4367. doi:10.1039/D0NA00539H.
- Leslie HA, van Velzen MJM, Brandsma SH, Vethaak AD, Garcia-Vallejo JJ, Lamoree MH. Discovery and quantification of plastic pollution in human blood. Environ Int. 2022; 107199. doi: 10.1016/j.envint.2022.107199.
- Lu L, Wan Z, Luo T, Fu Z, Jin Y. Polystyrene microplastics induce gut microbiota dysbiosis and hepatic lipid metabolism disorder in mice.Sci Total Environ. 2018; 631-632:449-458. doi: 10.1016/j.scitotenv.2018.03.051.
- Jin Y, Lu L, Tu W, Luo T, Fu Z. Impacts of polystyrene microplastic on the gut barrier, microbiota and metabolism of mice. Sci Total Environ. 2019; 649:308-317. doi: 10.1016/j.scitotenv.2018.08.353.
- Qiao J, Chen R, Wang M, […], Liu Y, Wu C, Chen C. Perturbation of gut microbiota plays an important role in micro/nanoplastics-induced gut barrier dysfunction. Nanoscale. 2021; 13:8806-8816. doi: 10.1039/d1nr00038a.
- Wilck N, Matus MG, Kearney SM, […], Linker RA, Alm EJ, Muller DN. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017; 551:585-589. doi: 10.1038/nature24628.
- Bartolomaeus H, Balogh A, Yakoub M, […], Muller DN, Stegbauer J, Wilck N. Short-Chain Fatty Acid Propionate Protects From Hypertensive Cardiovascular Damage. Circulation. 2019; 139:1407-1421. doi: 10.1161/CIRCULATIONAHA.118.036652.