
Principal Investigator

Dr. Christine K. Wong
Scientific interest within the context of the graduate college
Our airways are covered by a thin mucus layer that plays a pivotal role in maintaining lung health and homeostasis. In healthy, this mucus layer entraps constantly inhaled pathogens, pollutants and irritants that are then removed from the lungs by beating cilia on airway surfaces that facilitate mucocillary clearance, which constitutes an important innate defense mechanism of the lung. In chronic muco-obstructive lung diseases such as cystic fibrosis (CF) and bronchiectasis the viscoelastic properties of airway mucus are characteristically altered.1,2 Increased viscosity and elasticity of the mucus in these diseases leads to impaired mucociliary clearance, which in turn leads to airway mucus plugging, chronic infection with Pseudomonas aeruginosa and other bacterial pathogens, and chronic neutrophilic inflammation. Neutrophils express a group of proteases called neutrophil serine proteases (NSP), including neutrophil elastase (NE), protease 3 (PR3) and cathepsin G (CG). It is well established that increased activity of these NSPs leads to the degradation of endogenous anti-proteases, which in turn causes a protease/anti-protease imbalance that plays a central role in the pathogenesis of progressive structural lung damage in CF and bronchiectasis.3,4 Preliminary data from our group suggest that these proteases may also change the viscoelastic properties and function of the mucus by cleavage of the mucins (MUC5B and MUC5AC) that form the mucus layer. However, a systemic evaluation of the effects of NSPs on mucus properties has not been performed and whether proteolytic degradation of mucus is beneficial or detrimental in muco-obstructive lung diseases remains unknown. Finding answers to these questions is also relevant in the context of current development of novel therapies for CF and bronchiectasis that inhibits NSP activity5 and may thereby also have an effect on mucus properties and mucociliary clearance.
Project description
The overall aim of this project is to characterize the impact of NSPs on mucus properties and function in muco-obstructive lung diseases such as in CF and bronchiectasis. Elevated levels of NSP activity are characteristically found in expectorated sputum from patients with CF and patients with bronchiectasis,1,5 and are associating with increased mucus viscoelasticity and increased proinflammatory cytokines. Three NSPs have been identified, NE, PR3 and CG that have been implicated to play a crucial role in the pathogenesis of muco-obstructive lung diseases.6,7 To study the role of the three NSPs in mucin processing and modulation of mucus properties and function in health and disease, we will perform ex vivo studies of native sputum samples from healthy people and patients with CF or bronchiectasis and in vitro studies of the native mucus layer on patient-derived airway epithelial cultures. Specifically, sputum collected from CF or bronchiectasis and healthy people will be treated with individual NSPs (NE, PR3 or CG) at various pathophysiologically relevant concentrations and the impact on mucus processing and viscoelastic properties will be determined biochemically by Western blotting and mass spectrometry and functionally by rheological measurements using a cone and plate rheometer. Since NSP activity is commonly elevated in expectorated patient sputum, these measurements will also be performed in samples that will be treated with protease inhibitors. To gain mechanistic insight, these studies in patient sputum will be complemented by studies of highly differentiated patient-derived primary airway epithelial cell cultures that allow to study the native mucus layer under near physiological condition.8 For this purpose, airway cultures will be treated with the different NSPs added at different concentrations to the mucus layer and effects on viscoelastic properties will be determined using several sophisticated imaging-based techniques such as FRAP (fluorescent recovery after photobleaching)9 and magnetic microwire rheometry (MMWR)10 that have been established in our laboratory. By stimulating primary airway cultures with proinflammatory mediators in the presence of NSPs, we can recapitulate the chronic muco-inflammatory environment characteristic of CF and bronchiectasis in vitro. These functional studies of the properties of the native mucus layer will be accompanied by biochemical studies of abundancy and processing of the airway mucins MUC5AC and MUC5B by Western blotting and mass spectrometry, as described for patient sputum samples above, and inflammatory mediators such as proinflammatory cytokines secreted by epithelial cells will be assessed by bead-based multiplexed immunoassay. Thereby, this MD thesis will apply a spectrum of molecular, cellular, and state-of-the-art imaging techniques to patient-derived sputum and airway cultures to tackle pertinent questions related to the pathogenesis and therapy of muco-obstructive lung diseases such as in CF and bronchiectasis, which have emerged as the third leading chronic lung disease worldwide with limited therapeutic options.
References
- Schaupp L, Addante A, Völler M, Fentker K, Kuppe A, Bardua M, Duerr J, Piehler L, Röhmel J, Thee S, Kirchner M, Ziehm M, Lauster D, Haag R, Gradzielski M, Stahl M, Mertins P, Boutin S, Graeber SY, Mall MA. Longitudinal effects of elexacaftor/tezacaftor/ivacaftor on sputum viscoelastic properties, airway infection and inflammation in patients with cystic fibrosis. Eur Respir J. 2023; 62(2): 2202153.
- Ramsey KA, Chen ACH, Radicioni G, Lourie R, Martin M, Broomfield A, Sheng YH, Hasnain SZ, Radford-Smith G, Simms LA, Burr L, Thornton DJ, Bowler SD, Livengood S, Ceppe A, Knowles MR, Noone PG Sr, Donaldson SH, Hill DB, Ehre C, Button B, Alexis NE, Kesimer M, Boucher RC, McGuckin MA. Airway mucus hyperconcentration in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2020; 201(6): 661-670.
- Chalmers JD, Mall MA, Chotirmall SH, O’Donnell AE, Flume PA, Hasegawa N, Ringshausen FC, Watz H, Xu JF, Shteinberg M, McShane PJ. Targeting neutrophil serine proteases in bronchiectasis. Eur Respir J. 2025; 65(1): 2401050.
- Mall MA, Davies JC, Donaldson SH, Jain R, Chalmers JD, Shteinberg M. Neutrophil serine proteases in cystic fibrosis: role in disease pathogenesis and rationale as a therapeutic target. Eur Respir Rev. 2024; 33(173): 240001.
- Johnson ED, Long MB, Perea L, Shih VH, Fernandez C, Teper A, Cipolla D, McIntosh E, Galloway R, Eke Z, Shuttleworth M, Hull R, Spinou A, De Soyza A, Ringshausen FC, Goeminne P, Lorent N, Haworth C, Loebinger MR, Blasi F, Shteinberg M, Aliberti S, Polverino E, Sibila O, Shoemark A, Mange K, Huang JTJ, Stobo J, Chalmers JD. Broad immunomodulatory effects of the dipeptidyl-peptidase-1 inhibitor Brensocatib in bronchiectasis: data from the phase 2, double-blind, placebo-controlled WILLOW trial. Am J Respir Crit Care Med. 2025; 211(5): 770-778.
- Voynow JA, Shinbashi M. Neutrophil elastase and chronic lung disease. Biomolecules. 2021; 11(8): 1065.
- Clancy, D.M., et al. Extracellular neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep. 2018; 22(11): 2937-2950.
- Balázs A, Millar-Büchner P, Mülleder M, Farztdinov V, Szyrwiel L, Addante A, Kuppe A, Rubil T, Drescher M, Seidel K, Stricker S, Eils R, Lehmann I, Sawitzki B, Röhmel J, Ralser M, Mall MA. Age-related differences in structure and function of nasal epithelial cultures from healthy children and elderly people. Front Immunol. 2022; 13: 822437.
- Balázs A, Rubil T, Wong CK, Berger J, Drescher M, Seidel K, Stahl M, Graeber SY, Mall MA. The potentiator ivacaftor is essential for pharmacological restoration of F508del-CFTR function and mucociliary clearance in cystic fibrosis. JCI Insight. 2025; 10(10): e187951.
- Braunreuther M, Liegeois M, Fahy JV, Fuller GG. Nondestructive rheological measurements of biomaterials with a magnetic microwire rheometer. J Rheol. 2023; 67(2): 579-588.
Principle Investigator

Scientific interest within the context of the graduate college
This project aligns with Re-Thinking Health by investigating homeostatic immune control of a common virus, the John Cunningham virus (JCV), by deciphering the identity and function of rare populations of protective T-cells in preventing JCV reactivation and progressive multifocal leukoencephalopathy (PML). Up to 80-90% of all adults are infected with JCV, but the infection rarely causes disease. In contrast, individuals with compromised T cell immunity are at risk of PML, with high mortality and limited treatment options. Resolving antigenic targets and functional identity of protective T-cell responses, will enable design of novel vaccines and enable a more precise risk prediction in at-risk populations, such as patients under immune suppressive therapies. The program’s focus on health as an active regulated biological state, together with its emphasis on prevention-oriented translational research, provides an ideal framework to investigate adaptive immune mechanisms underlying latent viral control and maintenance of organismal health.
Project description
Human polyomaviruses are a family of DNA viruses that primarily establish persistent asymptomatic infections but can reactivate into virulent archetypes under conditions of immunosuppression. One such example is JCV, which exhibits tropism for oligodendrocytes in the central nervous system (CNS) upon reactivation, leading to the development of PML. PML is a rare but often fatal demyelinating disease of the CNS, with no licensed treatment options available to-date. Interventions including immune checkpoint inhibitors have yielded heterogenous response rates, and restoration of JCV-specific T-cell immunity currently remains the single most effective strategy to control CNS viral load and PML. Yet, this is not always possible, leaving a high unmet clinical need to better treat this debilitating disease.
While recent advances have opened promising opportunities for treatment of active PML, such as immune checkpoint blockade or adoptive T-cell transfer, our basic understanding of antigen specificity and functional capacity of protective T-cell immunity against PML remains surprisingly limited. This gap is largely attributable to technical challenges associated with detecting JCV-specific T-cells, which are present at exceptionally low frequencies in the periphery of healthy individuals, despite effective immune control. This hinders effective design of predictive assays for individuals under immunosuppression and also represents a barrier to identify protective T cells, TCRs, and their cognate epitopes profile; thus, posing a major roadblock in preventing and treating JCV reactivation and disease (PML).
To address this, we have previously i) developed a highly sensitive assay to detect rare JCV-specific CD4 T-cells from donor and patient derived peripheral blood, and ii) established a platform to isolate antigen-specific T-cell clones, and analyze their TCR and the cognate peptide epitopes, and extract transcriptomic signatures at single cell resolution. We are currently validating a sizeable set of CD4 TCR and peptide pairs. In the proposed project, we aim to extend this methodology to identify CD8 T cells, their TCRs, and MHC-I restricted epitopes. This will not only yield critical insights into JCV immune control, but it will provide a blueprint to study T cell dependent immune control to chronic viruses and build valuable datasets for machine learning based prediction of TCR – peptide pairing.
Aim 1. Adapt and validate T cell detection assay of JCV-specific CD8 T-cells.
Aim 2. Uncover JCV-specific CD8 T-cell clones, TCRs, their transcriptional profile and their cognate epitopes.
Aim 3. Validate the identified viral epitopes and TCRs by re-expression of TCRs.
Taken together, the proposed project will shed new light on immune control of JCV. Uncovered T-cell epitopes and TCRs present an attractive basis for the development of patient-specific cellular precision therapies.
References
- Jelcic I, Jelcic I, Faigle W, Sospedra M, Martin R. Immunology of progressive multifocal leukoencephalopathy. J. Neurovirol. 2015; 21(6): 614-622.
- Cortese I, Reich DS, Nath A. Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat Rev Neurol. 2021; 17(1): 37-51.
- Schulze Lammers FC, Bonifacius A, Tischer-Zimmermann S, Goudeva L, Martens J, Lepenies B, von Karpowitz M, Einecke G, Beutel G, Skripuletz T, Blasczyk R, Beier R, Maecker-Kolhoff B, Eiz-Vesper B. Antiviral T-cell frequencies in a healthy population: reference values for evaluating antiviral immune cell profiles in immunocompromised patients. J Clin Immunol. 2022; 42(3): 546-558.
- Pai JA, Satpathy AT. High-throughput and single-cell T cell receptor sequencing technologies. Nat Methods. 2021; 18(8): 881-892.
- Georg P, Astaburuaga-García R, Bonaguro L, Brumhard S, Michalick L, Lippert LJ, Kostevc T, Gäbel C, Schneider M, Streitz M, Demichev V, Gemünd I, Barone M, Tober-Lau P, Helbig ET, Hillus D, Petrov L, Stein J, Dey HP, Paclik D, Iwert C, Mülleder M, Aulakh SK, Djudjaj S, Bülow RD, Mei HE, Schulz AR, Thiel A, Hippenstiel S, Saliba AE, Eils R, Lehmann I, Mall MA, Stricker S, Röhmel J, Corman VM, Beule D, Wyler E, Landthaler M, Obermayer B, von Stillfried S, Boor P, Demir M, Wesselmann H, Suttorp N, Uhrig A, Müller-Redetzky H, Nattermann J, Kuebler WM, Meisel C, Ralser M, Schultze JL, Aschenbrenner AC, Thibeault C, Kurth F, Sander LE, Blüthgen N, Sawitzki B; PA-COVID-19 Study Group. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell. 2022; 185(3): 493-512.e25.
Prinicipal Investigator

Dr. István András Szijarto, PhD
Scientific interest within the context of the graduate college
Scientific interest centers on the molecular mechanisms that preserve health and resilience with focus on microbiome-host crosstalk, especially microbiome-derived hydrogen sulfide, protein persulfidation, and systemic energy metabolism in organ function. This aligns with the goals of Re-Thinking Health, which aims to define health-preserving pathways and translate them into prevention-oriented strategies.
Project description
Our research group is investigating the role of the microbiome in kidney and cardiovascular diseases.1,2 Interestingly, patients with kidney disease face a substantial cardiovascular risk.3 An altered microbiome may contribute to an increased cardiovascular risk in the presence of existing kidney dysfunction.4 However, the exact mechanisms remain poorly understood, leading to a lack of therapeutic options. We are particularly interested in the molecules that mediate the interaction between the microbiome and host cells.5-9
Hydrogen sulfide (H2S) is best known as the gas that smells like rotten eggs, yet in the body it also acts as an important signaling molecule.10 It can modify proteins through persulfidation, influence mitochondrial energy production, and shape inflammatory responses. These functions are highly relevant to common chronic diseases such as heart failure and chronic kidney disease. The intestinal microbiota is a major source and regulator of H2S, but we still do not know how microbiome-derived H2S reaches distant organs, how it changes tissue function, or whether this pathway can be harnessed therapeutically.
Our hypothesis is that microbiome-derived H2S supports systemic resilience by maintaining organ-specific persulfidation patterns and efficient cellular energy metabolism. Loss or dysregulation of this pathway increases susceptibility to heart, kidney, and immune dysfunction, particularly under stress. This proposal aims to define the mechanistic link between gut microbial H2S production and remote organ function in a biologically innovative and clinically relevant manner.
Aim 1: Define how the microbiome regulates systemic H2S. We will identify how the microbiome contributes to systemic H2S homeostasis. Using germ-free and colonized mice, together with human microbiome samples, we will profile microbial pathways involved in H2S production by shotgun sequencing and targeted qPCR. We will then quantify H2S levels, key precursors, and protein persulfidation across the gut and distant organs to test whether a measurable gut-to-organ gradient exists. In parallel, we will assess host H2S -producing and H2S -metabolizing enzymes to determine whether the microbiome also alters host H2S synthesis or breakdown.
Aim 2: Test how microbial H2S shapes remote organ resilience. We will test how microbiome-derived H2S affects the heart, kidney, immune system, and intestine. We propose that reduced microbial H2S alters the persulfidome and weakens mitochondrial function in energy-demanding tissues. Germ-free and colonized mice will be compared at baseline and in a stress model of heart failure with kidney injury. Organ phenotypes will be linked to bioenergetics, inflammation, and tissue persulfidation, while rescue experiments with an oral H2S donor will test causality.
Aim 3: To strengthen translational relevance. We will assess the H₂S-producing capacity of the microbiota in patients with end-stage kidney disease compared to healthy controls to determine whether modulation of microbial H₂S production could serve as a novel strategy to enhance resilience and limit organ damage.
References
- Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Markó L, Vvedenskaya O, Kleiner FH, Tsvetkov D, Klug L, Costea PI, Sunagawa S, Maier L, Rakova N, Schatz V, Neubert P, Frätzer C, Krannich A, Gollasch M, Grohme DA, Côrte-Real BF, Gerlach RG, Basic M, Typas A, Wu C, Titze JM, Jantsch J, Boschmann M, Dechend R, Kleinewietfeld M, Kempa S, Bork P, Linker RA, Alm EJ, Müller DN. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017; 551(7682): 585-589.
- Holle J, Bartolomaeus H, Löber U, Behrens F, Bartolomaeus TUP, Anandakumar H, Wimmer MI, Vu DL, Kuhring M, Brüning U, Maifeld A, Geisberger S, Kempa S, Schumacher F, Kleuser B, Bufler P, Querfeld U, Kitschke S, Engler D, Kuhrt LD, Drechsel O, Eckardt KU, Forslund SK, Thürmer A, McParland V, Kirwan JA, Wilck N, Müller D. Inflammation in children with CKD linked to gut dysbiosis and metabolite imbalance. J Am Soc Nephrol. 2022; 33(12): 2259-2275.
- Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. New Engl J Med. 2004; 351(13): 1296-1305.
- Bloom PP, Garrett WS, Penniston KL, Winkler MH, Hazen SL, Agudelo J, Suryavanshi M, Babiker A, Dodd D, Fischbach MA, Huang KC, Huttenhower C, Joe B, Kalantar-Zadeh K, Knight R, Miller AW, Rabb H, Srivastava A, Tang WHW, Turnbaugh PJ, Walker AW, Wilck N, Xu J, Yang T, Himmelfarb J, Redinbo MR, Wu GD, Woodworth MH, Ackerman AL, Winter S, Rinschen MM, Hassan HA, Biruete A, Anderson AH, Pluznick JL. Microbiota and kidney disease: the road ahead. Nat Rev Nephrol. 2025; 21(10): 702-716.
- Yarritu A, Anders W, Thiele A, Potapenko O, Schumacher F, Szijártó IA, Matz-Rauch A, Versnjak J, Gebremedhin N, McParland V, Heckscher S, Kamboj S, Trimarchi G, Anandakumar H, Fuckert F, Hoffmann C, Hassan SA, Bonnekoh PM, Wimmer MI, Behrens F, Voelkl J, Kramann R, Kintscher U, Kuehne T, Kleuser B, Zernecke A, Oefner PJ, Gronwald W, Dettmer K, Eckardt KU, Müller DN, Kelm M, Holle J, Bartolomaeus H, Wilck N. Kidney disease reprograms microbiome-host signaling to promote heart failure. bioRxiv. 2026. 2026.2001.2010.698142.
- Wimmer MI, Reichel M, Thiele A, Yarritu A, Matz-Rauch A, Anandakumar H, Götz LH, Lesker TR, Potapenko O, Gebremedhin N, Anders W, Liévano Contreras SV, Wang R, Nonn O, Schiattarella GG, Schaefer F, Holle J, Strowig T, Zernecke A, Eckardt KU, Knauf F, Wilck N, Bartolomaeus H. Interleukin-17A mediates cardiorenal injury in oxalate nephropathy. bioRxiv. 2025. 2025.2011.2017.687153.
- Wimmer MI, Bartolomaeus H, Anandakumar H, Chen CY, Vecera V, Kedziora S, Kamboj S, Schumacher F, Pals S, Rauch A, Meisel J, Potapenko O, Yarritu A, Bartolomaeus TUP, Samaan M, Thiele A, Stürzbecher L, Geisberger SY, Kleuser B, Oefner PJ, Haase N, Löber U, Gronwald W, Forslund-Startceva SK, Müller DN, Wilck N. Metformin modulates microbiota and improves blood pressure and cardiac remodeling in a rat model of hypertension. Acta Physiol (Oxf). 2024; 240(11): e14226.
- Bartolomaeus H, Balogh A, Yakoub M, Homann S, Markó L, Höges S, Tsvetkov D, Krannich A, Wundersitz S, Avery EG, Haase N, Kräker K, Hering L, Maase M, Kusche-Vihrog K, Grandoch M, Fielitz J, Kempa S, Gollasch M, Zhumadilov Z, Kozhakhmetov S, Kushugulova A, Eckardt KU, Dechend R, Rump LC, Forslund SK, Müller DN, Stegbauer J, Wilck N. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation. 2019; 139(11): 1407-1421.
- Bartolomaeus H, Avery EG, Bartolomaeus TUP, Kozhakhmetov S, Zhumadilov Z, Muller DN, Wilck N, Kushugulova A, Forslund SK. Blood pressure changes correlate with short-chain fatty acid production potential shifts under a synbiotic intervention. Cardiovasc Res. 2020; 116(7): 1252-1253.
- Filipovic MR, Zivanovic J, Alvarez B, Banerjee R. Chemical Biology of H2S Signaling through Persulfidation. Chem Rev. 2018; 118(3): 1253-1337.
Principal Investigator

Scientific interest within the context of the graduate college
Rheumatoid Arthritis (RA) represent a chronic and prevalent autoimmune disease characterized by inflammation and progredient joint destruction. Both anti-citrullinated protein antibodies (ACPA) and autoreactive ACPA-producing B cells play a key role in disease pathogenesis. However, the exact mechanisms leading to disease onset remain poorly understood. Notably, ACPA are not only present in RA patients, but can be also detected in a certain fraction of healthy individuals where they are indicating an increased risk of developing RA in the future. The proposed study thus aims to investigate the role of autoreactive ACPA-producing B cells in ACPA-positive healthy individuals and to understand the molecular mechanisms that eventually lead to the onset of inflammatory disease. By focusing on autoreactive B cells, we aim to uncover novel molecular insights into the pathogenesis of RA and identify potential therapeutic targets to prevent disease progression.
Project description
We have collected a cohort of ACPA-positive healthy individuals as well as of ACPA-positive RA patents. The project aims to perform a molecular phenotyping of B cells in these patients where we seek to understand the behavior, characteristics and differences of autoreactive cells in ACPA-positive healthy individuals and RA patients. We plan to apply cutting edge technology including bar-coded tetramers, mass cytometry and single cell sequencing to characterize autoreactive ACPA-producing B cells. The overall aim is to understand the mechanisms that lead to onset of disease.
Aim 1: Examine the phenotypic and functional characteristics of autoreactive B cells in ACPA-positive individuals and RA patients using mass cytometry. We will use mass cytometry to perform a deep phenotyping of PBMCs of ACPA-positive individuals and RA patients where we will focus on activation markers and signaling pathway activity in response to ex vivo stimulation.
Aim 2: Examine the phenotypic and functional characteristics of autoreactive B cells in ACPA-positive individuals and RA patients using scRNAseq. Using scRNAseq and bar-coded tetramers, we will focus on the molecular phenotype of ACPA-producing autoreactive B cells in ACPA-positive individuals and RA patients.
Aim 3: Clinical Correlation. Access to clinical datasets will allow assessment of the correlation between B cell characteristics and clinical outcomes in ACPA-positive individuals.
References
- Schett G, Nagy G, Krönke G, Mielenz D. B-cell depletion in autoimmune diseases. Ann Rheum Dis. 2024; 83(11): 1409-1420.
- Wilhelm A, Chambers D, Müller F, Bozec A, Grieshaber-Bouyer R, Winkler T, Mougiakakos D, Mackensen A, Schett G, Krönke G. Selective CART cell-mediated B cell depletion suppresses IFN signature in SLE. JCI Insight. 2024; 9(12): e179433.
- Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, Völkl S, Simon D, Kleyer A, Munoz L, Kretschmann S, Kharboutli S, Gary R, Reimann H, Rösler W, Uderhardt S, Bang H, Herrmann M, Ekici AB, Buettner C, Habenicht KM, Winkler TH, Krönke G, Schett G. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. 2022; 28(10): 2124-2132.
- Scherer HU, Huizinga TWJ, Krönke G, Schett G, Toes REM. The B cell response to citrullinated antigens in the development of rheumatoid arthritis. Nat Rev Rheumatol. 2018; 14(3): 157-169.
- Pfeifle R, Rothe T, Ipseiz N, Scherer HU, Culemann S, Harre U, Ackermann JA, Seefried M, Kleyer A, Uderhardt S, Haugg B, Hueber AJ, Daum P, Heidkamp GF, Ge C, Böhm S, Lux A, Schuh W, Magorivska I, Nandakumar KS, Lönnblom E, Becker C, Dudziak D, Wuhrer M, Rombouts Y, Koeleman CA, Toes R, Winkler TH, Holmdahl R, Herrmann M, Blüml S, Nimmerjahn F, Schett G, Krönke G. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat Immunol. 2017; 18(1): 104-113.
Principle Investigator

Scientific interest within the context of the graduate college
The epithelium of the gastrointestinal tract is a single cell layer organized into villi and crypts, which separates microbiota and host. Stem cells in the crypts constantly generate epithelial progenitors that differentiate and migrate toward the villus tip, where they are shed into the lumen. While some of these processes are epithelial-intrinsic, coordinated interactions with underlying stromal cells are essential for tissue morphogenesis, stem cell maintenance, epithelial differentiation and spatial zonation along the crypt–villus axis. Importantly, disruption of this communication not only impairs local tissue integrity but also drives systemic inflammation, metabolic dysfunction, and cancer risk. Understanding these interactions will provide key insights into the mechanisms linking tissue-specific processes to overall organismal health and disease.
In our lab, we specifically investigate how intestinal stromal and epithelial cells communicate to maintain tissue homeostasis. Recently, we identified the transcription factor c-Maf as a key regulator of enterocyte differentiation controlled by stromal Noggin/BMP signals, linking spatial patterning along the villus axis to nutrient absorption and epithelial function. Disruption of this pathway impairs enterocyte maturation and alters immune and microbial homeostasis. Our current work focuses on the molecular pathways, cellular interactions, and spatial organization that coordinate stromal–epithelial crosstalk within the gut microenvironment. By integrating cell biology, genomics, and advanced imaging approaches, we aim to define how these interactions are perturbed in intestinal disease and to identify strategies to restore intestinal function.
Project description
In the healthy gut, the delicate balance between intestinal epithelial cell (IEC) proliferation, differentiation and apoptosis maintains barrier integrity, however, chronic inflammation disrupts this homeostasis. While stromal cells are known to regulate IEC fate via niche-derived signals, the specific molecular mediators governing these interactions remain incompletely defined. Our preliminary work identifies the opioid growth factor (OGF) and its receptor (OGFR) pathway as a previously unrecognized signalling axis between intestinal stromal cells and IECs, that is markedly upregulated during gut inflammation. Based on these findings, this project aims to comprehensively characterize this crosstalk, its regulation and functional relevance for IEC and gut homeostasis and inflammation.
To define the signals that regulate intestinal OGF/OGFR expression. While IECs are known to express OGFR and specific stromal cell populations produce OGF, the upstream signals controlling their expression remain poorly understood. Based on preliminary data, we hypothesize that homeostatic cues, including microbial- and immune cell-derived signals, regulate OGF/OGFR expression under steady-state conditions. Furthermore, our findings of increased OGF/OGFR expression during intestinal inflammation suggest that pro-inflammatory cytokines also contribute to their regulation. The objective of this project is to systematically identify and characterize the signals governing OGF/OGFR expression in the intestine.
Aim 1: Which signals regulate OGFR expression in intestinal IECs? 3D intestinal organoids (“gut in a dish”) are generated from primary mouse intestinal epithelial cells (IECs) using cell culture techniques. Cytokine stimulation experiments are then performed with these 3D intestinal organoids. Following stimulation, OGFR expression in the organoids is analyzed by quantitative PCR (qPCR). In addition, primary mouse IECs are isolated from different transgenic mouse strains (e.g., microbiota-deficient or immunodeficient mice) using fluorescence-activated cell sorting (FACS). Finally, endogenous OGFR expression in the FACS-sorted IECs is measured by qPCR.
Aim 2: Which signals regulate OGF expression in intestinal stromal cells? Primary mouse intestinal stromal cells are isolated using fluorescence-activated cell sorting (FACS). These cells are then cultured under appropriate cell culture conditions. Cytokine stimulation experiments are subsequently performed with the mouse intestinal stromal cells. Following stimulation, OGF expression in the stromal cells is analyzed by quantitative PCR (qPCR). In addition, gut tissue samples are prepared for immunofluorescence (IF) microscopy, including embedding, sectioning, and mounting. Finally, endogenous OGF expression is analyzed by IF microscopy in OGF reporter mice.
This project will combine different in vitro and in vivo approaches to define the signals that regulate OGFR and OGF expression in intestinal IECs and stromal cells, respectively. Key techniques will include isolation of primary mouse intestinal IECs and stromal cells, cell culture, 3D intestinal organoids, flow cytometry and flow cytometry-mediated cell sorting, tissue embedding, cryosectioning, staining, immunofluorescence microscopy and qPCR.
References
- Cosovanu C, Resch P, Jordan S, Lehmann A, Ralser M, Farztdinov V, Spranger J, Mülleder M, Brachs S, Neumann C. Intestinal epithelial c-Maf expression determines enterocyte differentiation and nutrient uptake in mice. J Exp Med. 2022; 219(12): e20220233.
Prinicipal Investigator
Scientific interest within the context of the graduate college
The goal of my laboratory is to understand the regulation and control mechanisms of immune responses at barrier surfaces specifically in the respiratory tract. We are continuously exposed to the environment by filtering liters of air each minute to provide needed oxygen in exchange to carbon dioxide to our body. A unique network of pulmonary immune cell populations ensures appropriate immune responses not only against pathogens or hazardous materials but to maintain lung function and health at steady state. However, the underlying mechanisms are poorly understood. Group 2 innate lymphoid cells (ILC2s) are the major innate lymphoid immune cell population in the lungs, tissue resident and able to orchestrate innate but also adaptive immune responses. Thus, studying the regulatory processes of ILC2 effector function is key to understand immune concepts of pulmonary immunity and health.
Project description
The microbiome plays a fundamental role in shaping the immune system and is a key determinant of health and disease. Microbial components and metabolites critically regulate ILC2 responses, influencing immune homeostasis and host defense. In this project, we will investigate how distinct microbiome compositions modulate immune cell function, with a particular focus on ILC2 responses in the lung during respiratory viral infection. A unique aspect of this study is the comparison of mice harboring a conventional laboratory microbiome with those colonized by a natural, “physiological” microbiome, enabling us to assess how microbiome diversity influences immune regulation and antiviral immunity. Overall, this project integrates immunological, microbiome, and advanced imaging approaches to provide mechanistic insights into host–microbiome interactions. By uncovering microbiome-dependent mechanisms that shape antiviral immunity, this work has the potential to inform novel microbiome-based therapeutic strategies to enhance immune protection and improve outcomes in respiratory viral infections.
Aim 1: To investigate how the host microbiome shapes the immune response to respiratory viral infections. Cellular and humoral components of the immune system will be analyzed at different timepoints with a focus on ILC2s. In addition, viral titer and lung histology will be determined. The analysis will include male vs female mice to determine and define differences based on biological sex in immune cell activation and effector function.
Aim 2: To study the composition and diversity of a physiological microbiome upon infection. The microbiome regulates pulmonary health and microbial components (in)directly regulate ILC2 function. The microbial composition will be determined during infection and analyzed by culture and sequencing analysis.
References
- Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, McCulloch JA, Anastasakis DG, Sarshad AA, Leonardi I, Collins N, Blatter JA, Han SJ, Tamoutounour S, Potapova S, Foster St Claire MB, Yuan W, Sen SK, Dreier MS, Hild B, Hafner M, Wang D, Iliev ID, Belkaid Y, Trinchieri G, Rehermann B. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science. 2019; 365(6452): eaaw4361.
- Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A. 2011; 108(13): 5354-5359.
- Oh JH, Hild B, Yoshida T, Badger JH, Seishima J, McCulloch JA, Jung MK, Azar S, Trinchieri G, Rehermann B. Naturalized immune responses are stable over years in a colony of laboratory mice with wild-derived microbiota. Immunity. 2025; 58(9): 2305-2319.e5.
- Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, Eva MM, Gauchat JF, Qureshi ST, Mazer BD, Mossman KL, Malo D, Gamero AM, Vidal SM, King IL, Sarfati M, Fritz JH. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. 2016; 17(1): 65-75.
Principal Investigator

Prof. Dr. Ildiko Dunay
Scientific interest within the context of the graduate college
Our lab investigates fundamental mechanisms of communication between the nervous and immune systems, two major sensory networks that continuously monitor tissue integrity and initiate protective responses upon disruption. While the immune system’s role in maintaining barrier function is increasingly understood, the contribution of neuronal signals to immune regulation remains largely unexplored. Recent work from the lab has revealed how neuropeptides such as neuromedin U and neurotransmitters like norepinephrine modulate innate lymphoid cells (ILCs), key tissue-resident immune cells at mucosal barriers. By integrating advanced genetic tools with state-of-the-art methods from immunology, neuroscience, and genomics, the lab aims to dissect the cellular and molecular circuits of neuro-immune interaction.
Project description
Toxoplasma gondii, a ubiquitous intracellular parasite with a profound ability to infect virtually all warm-blooded animals, is responsible for a significant public health concern. In humans, infection can lead to toxoplasmosis, which, while often asymptomatic in healthy individuals, poses severe risks to immunocompromised patients and pregnant women.1 Moreover, emerging evidence suggests T. gondii’s potential triggering of various neurological disorders, including schizophrenia and other psychiatric conditions, underscoring the critical need to understand the parasite’s interaction with the nervous system.2 The central and enteric nervous systems are intricate networks that regulate numerous vital functions, from cognition and sensory processing in the central nervous system (CNS) to gastrointestinal motility and secretion in the enteric nervous system (ENS).3 T. gondii’s ability to invade and persist in these neural environments suggests significant adaptations by both the parasite and host neurons. Investigating these adaptations, particularly at the transcriptional level, provides insights into the mechanisms of pathogenesis, neuronal resilience, and potential neuropathology associated with infection.
We aim to monitor the transcriptional changes occurring in neurons during T. gondii infection. To this end, we have already established the respective lines and protocols for bulk sequencing of purified RNA from sort-purified GFP+ neuronal nuclei from the T. gondii-infected brain and gut tissue of the Snap25Cre/+ x INTACT system, in which the GFP expression is only activated in neurons. To monitor alterations in enteric neurons’ composition and transcriptional changes in subsets of enteric neurons, we will perform RNA sequencing from brains and gut tissue. To this end, we will separately sort-purify GFP+ nuclei from Snap25Cre/+ x INTACT system to obtain highly pure neuronal nuclei. These neuronal cell nuclei will be analyzed using the RNA-sequencing platform, which we have already implemented for single-nuclei sequencing. Together, these experiments will identify genes in neurons regulated in the context of T. gondii infection.
Based on our data detecting a type II interferon signature in the ENS in various inflammation models, we will perform bulk RNA sequencing of the ENS and CNS, in which we have deleted the Ifngr1 specifically in neurons by the Snap25Cre/+ x Ifngr1fl/fl INTACT system and which allow for sort-purification based on the nuclear GFP signal. This experimental setup will allow us to study the potentially protective gene expression signature in neurons induced by IFN-γ. Furthermore, these experiments will enable us measure if neuron-specific deletion of the IFN-γ receptor results in increased T.gondii burden in various organs. Therefore, these experiments will reveal how IFN-γ-signaling in neurons contributes to anti-toxoplasma immunity.
Taken together, these datasets will yield correlations of differentially expressed genes of ENS and CNS neurons and their relative abundances, exposing disease-relevant signaling circuits triggered in neurons. This global perspective is essential for uncovering the molecular underpinnings of T. gondii’s impact on neuronal function and survival, which could lead to the development of targeted therapies and interventions.1,2
References
- Dunay IR, Gajurel K, Dhakal R, Liesenfeld O, Montoya JG. Treatment of toxoplasmosis: historical perspective, animal models, and current clinical practice. Clin Microbiol Rev. 2018; 31(4): e00057-17.
- Matta SK, Rinkenberger N, Dunay IR, Sibley LD. Toxoplasma gondii infection and its implications within the central nervous system. Nat Rev Microbiol. 2021; 19(7): 467-480.
- Jakob MO, Kofoed-Branzk M, Deshpande D, Murugan S, Klose CSN. An integrated view on neuronal subsets in the peripheral nervous system and their role in immunoregulation.Front Immunol. 2021; 12: 679055.
Principal Investigator

Dr. Lasti Erfinanda
Scientific interest within the context of the graduate college
Pulmonary hypertension is a common cardiopulmonary disease that is characterized by extensive remodeling of the pulmonary vascular tree and a poor prognosis due to ultimate right heart failure. The underlying progressive lung vascular remodeling involves two distinct but interconnected pathologies: distal capillary loss (vascular rarefaction) and proliferative remodeling of precapillary resistance vessels. Current therapeutic approaches rely on vasodilatory drugs and do not address the underlying causes of vascular remodeling which are poorly understood. Yet, deeper insight into the underlying pathophysiological processes – which forms the basis for the development of causal rather than symptomatic therapies – is hampered by limited access to human biosamples, the restriction to post-mortem end-point analyses in animal models, and the complexity of the multicellular processes driving vascular remodeling which cannot be recapitulated in traditional cell culture. To overcome this gap in methodologies, knowledge and ultimately therapy, we have engaged in a collaboration with a Suisse bioengineering lab to develop in-vitro microvasculature-on-chip and artery-on-chip models that allow for the first time to track changes of the vascular system and the dynamics of individual cell types in an unprecedented temporally and spatially resolved context.1 These models – in combination with advanced imaging modalities, state-of-the-art metabolic assays and functional read-outs – are now ready to use and open up unprecedented avenues for the discovery of new mechanisms of health and disease and the development of new therapeutic targets.
Project description
Pulmonary hypertension is an ultimately fatal disease characterized by an increase in pulmonary vascular resistance that results from a combination of capillary rarefaction and inward remodeling of pulmonary arteri(ol)es.2 Recently, pericytes have been implicated in both of these events: Pericytes are perivascular cells that are encased within the microvascular basement membrane and assist in the maturation and stabilization of microvascular networks. To this end, pericytes closely communicate with endothelial cells promoting the stabilization of endothelial cells and the maintenance of vascular barrier function. In vitro, pericytes can even “donate”, i.e. transfer mitochondria to endothelial cells via so-called tunnelling nanotubes (TNTs).3
In pulmonary hypertension, however, pericytes detach from capillary networks, possibly contributing to their disintegration.4 In parallel, pericytes emerge at the level of pulmonary arteries where they integrate into the muscular layer of the vascular wall likely contributing to medial thickening.5 In previous work, we have shown i) that pericytes form TNTs with lung microvascular endothelial cells in intact lung capillary networks, ii) that these interaction sites show abundant expression of connexin 43, a gap junctional molecule known to coordinate mitochondrial transfer between cells,6 and iii) that pulmonary artery endothelial cells in pulmonary hypertension undergo a metabolic switch from oxidative phosphorylation to glycolysis (a phenomenon known from cancer cells as “Warburg-effect” that allows cancer cells to hyperproliferate). Based on these observations and published data demonstrating a similar metabolic switch in pericytes in pulmonary hypertension,7 we propose a paradigm-shifting metabolic concept for the co-occurrence of capillary loss and arteriolar remodeling in pulmonary hypertension that positions the pericyte in the center of the pathophysiology: In healthy homeostasis, pericytes closely interact with microvascular endothelial cells, supporting their oxidative phosphorylation by transferring mitochondria in a connexing 43-dependent manner to sustain capillary homeostasis. In pulmonary hypertension, pericytes undergo metabolic reprogramming toward increased glycolysis, resulting in their detachment from capillaries and migration to upstream resistance vessels where they integrate into the vascular wall, transferring glycolytic mitochondria to pulmonary artery smooth muscle cells thus in turn promoting their hyperproliferation and subsequent arteriolar remodeling while capillaries now disassemble in the absence of mitochondrial support from pericytes.
Our microvasculature-on-chip and artery-on-chip models in conjunction with classic co-culture systems are uniquely positioned to address this hypothesis via the following research aims:
Aim 1. To map mitochondrial transfer dynamics in lung capillary networks in health and pulmonary hypertension, we will use multi-color confocal and multi-photon imaging to track in real-time the interaction between pericytes and endothelial cells (identified by different fluorescent markers) and the transfer of fluorescently labelled mitochondria from one to another via TNTs. Experiments will be run in vascular networks formed by primary vascular cells from healthy donors and those from patients with pulmonary hypertension with the expectation that in the latter scenario, pericytes will detach from the capillaries and stop transferring mitochondria to lung microvascular endothelial cells, resulting in capillary disintegration and network rarefaction. Using dedicated assays for mitochondrial metabolism (Seahorse stress assays, Oroboros assays, metabolomic analyses by GC-MS) we will analyze the effects of this mitochondrial transfer (or lack thereof) on endothelial metabolism and function (in terms of proliferation versus apoptosis). The proposed regulatory role of connexin 43 will be tested using pharmacological inhibitors or enhancers of connexin 43-mediated gap junctional communication.
Aim 2. Using a newly engineered arteriole-on-a-chip platform, we will next probe whether pericytes with a pharmacologically or genetically induced glycolytic shift preferentially integrate into the arterial wall and (instead of promoting oxidative phosphorylation in lung capillaries) now start to donate glycolytic mitochondria to pulmonary artery smooth muscle cells, driving their proliferation. An analogous set of imaging, metabol(om)ic and functional assays will be applied as for Aim 1.
Aim 3. Finally, we will utilize a presently custom-designed platform combining a capillary network with a feeding arteriole on a single organ-on-a-chip to track the complete cascade of events from initial capillary homeostasis with quiescent pericytes to pericyte glycolytic switching, detachment and migration to the upstream arteriole where they ultimately trigger vascular remodeling by donating “injurious”, i.e. glycolytic mitochondria to smooth muscle cells.
The results of the proposed are expected to radically reshape our understanding of pulmonary hypertension, positioning changes in pericyte metabolism at the very center of the pathophysiology and identifying strategies to restore pericyte quiescence and homeostatic metabolism as a promising new avenue for the treatment of a presently uncurable disease.
References
- Bichsel CA, Hall SR, Schmid RA, Guenat OT, Geiser T. Primary human lung pericytes support and stabilize in vitro perfusable microvessels. Tissue Eng Part A. 2015; 21(15-16): 2166-2176.
- Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano-Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke-Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S; ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023; 61(1): 2200879.
- Kempf S, Popp R, Naeem Z, Frömel T, Wittig I, Klatt S, Fleming I. Pericyte-to-endothelial cell communication via tunneling nanotubes is disrupted by a diol of docosahexaenoic acid. Cells. 2024; 13(17): 1429.
- Yuan K, Shamskhou EA, Orcholski ME, Nathan A, Reddy S, Honda H, Mani V, Zeng Y, Ozen MO, Wang L, Demirci U, Tian W, Nicolls MR, de Jesus Perez VA. Loss of endothelium-derived Wnt5a is associated with reduced pericyte recruitment and small vessel loss in pulmonary arterial hypertension. Circulation. 2019; 139(14): 1710-1724.
- Bordenave J, Tu L, Berrebeh N, Thuillet R, Cumont A, Le Vely B, Fadel E, Nadaud S, Savale L, Humbert M, Huertas A, Guignabert C. Lineage tracing reveals the dynamic contribution of pericytes to the blood vessel remodeling in pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2020; 40(3): 766-782.
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012; 18(5): 759-765.
- Yuan K, Shao NY, Hennigs JK, Discipulo M, Orcholski ME, Shamskhou E, Richter A, Hu X, Wu JC, de Jesus Perez VA. Increased pyruvate dehydrogenase kinase 4 expression in lung pericytes is associated with reduced endothelial-pericyte interactions and small vessel loss in pulmonary arterial hypertension. Am J Pathol. 2016; 186(9): 2500-2514.
Principal Investigator

Scientific interest within the context of the graduate college
Our group aims to understand the molecular mechanisms involved in tissue homeostasis, inflammation, and resolution of inflammation. Our main focus is on the transcription factor NF-κB and its role in the intestinal epithelium. Our current projects range from determining the role of the transcription factor in epithelial regeneration in colitis and in inflammatory bowel diseases (Re-Thinking Health, 2022), to refining analgesia (Charité 3R), to investigating the role of NF-κB in cellular senescence in the gut (DFG) or its role in metabolism.
Project description
Senescent cells are characterized by terminal cell cycle arrest, epigenetic changes, and by the senescence associated secretory phenotype (SASP). SASP is comprised of scores of cytokines, chemokines, and growth factors, which regulate cells and tissues in a paracrine manner. Therefore, even a small number of senescent cells can have a dramatic impact on distant tissues and were shown to sustain inflammation, fuel carcinogenesis, and shorten lifespan. Most of the genes that code for SASP factors are transcriptionally regulated by NF-κB8.1,2 Under physiological conditions SASP promotes wound healing and recruitment of immune cells, which help clear senescent cells.
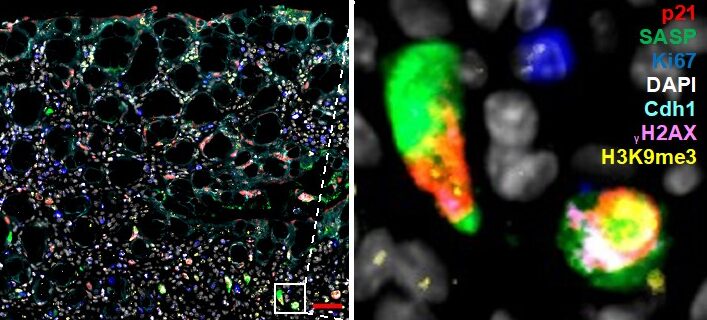
Figure 1. Multiplex immunofluorescence staining of senescence. Mouse colon (colitis model) was stained with markers for senescence. Simultaneous staining with numerous markers (multiplex) is required to rule out other cell states (proliferation, apoptosis, quiescence). Zoom-in on select fully senescent cells is shown on the right.
We are interested in investigating what kind of senescent populations arise in inflammatory bowel diseases (IBD) versus those that progressed to colorectal cancer (CRC). We are especially interested in the role that SASP plays in disease progression and whether distinct forms of SASP can be exploited therapeutically.
As part of our preliminary data, we have already identified distinct populations dependent on stage of inflammation and importantly, we show that no single type of senescence exists, but rather cells display different proficiencies for SASP and thus for communication with their environment. It is not known how these differences skew progression of IBD.
Aim 1: Characterize senescent cells in IBD and CRC and in murine colitis and CRC models. As part of this work package you will perform multiplex stainings (as in Figure 1) using already available samples from IBD and CRC patient samples and from murine mouse models. You will also receive training in basic analysis of single-cell RNA sequencing and will therefore quantify cells that have SASP versus those that do not. You will then isolate these cells using FACS and determine how different types of SASP alter immune cell recruitment.
Aim 2: Reprogram senescent cells to transform immunosuppressive “cold” tumor microenvironment environment into “hot”. Here you will use available senolytics and senomorphics (drugs that kill senescent cells or suppress their SASP), and those identified via our collaborators through deep learning, to reprogram senescent cells in 2D culture and in human and mouse intestinal organoids. Successful senolytics will be used in pre-clinical trials in CRC mouse models to determine if these expose tumors to immunosurveillance.
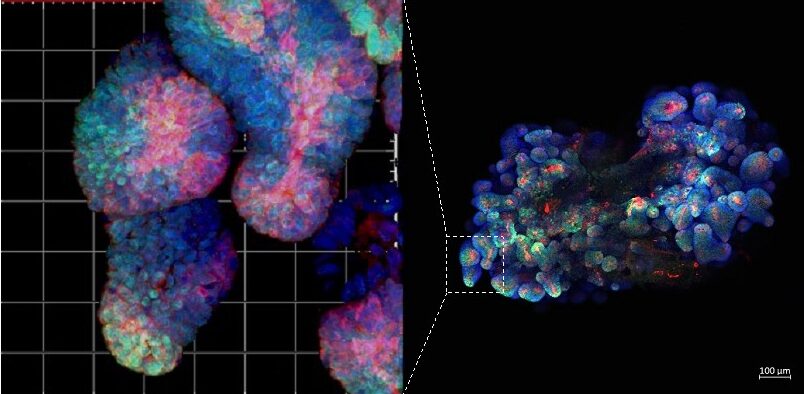
Figure 2. Immunofluorescence staining of mouse fluorescent organoid. All our projects combine work with mouse models. patient samples (including organoids) and bioinformatics.
Methods you will learn as part of this project: multiplex immunofluorescent staining, single-cell data analysis, immunohistochemistry, human and mouse organoid cultures (Figure 2), qPCR.
References
- Kolesnichenko M, Mikuda N, Höpken UE, Kärgel E, Uyar B, Tufan AB, Milanovic M, Sun W, Krahn I, Schleich K, von Hoff L, Hinz M, Willenbrock M, Jungmann S, Akalin A, Lee S, Schmidt-Ullrich R, Schmitt CA, Scheidereit C.. Transcriptional repression of NFKBIA triggers constitutive IKK- and proteasome-independent p65/RelA activation in senescence. EMBO J. 2021; 40(6):e104296.
- Mikuda N, Schmidt-Ullrich R, Kärgel E, Golusda L, Wolf J, Höpken UE, Scheidereit C, Kühl AA, Kolesnichenko M. Deficiency in IκBα in the intestinal epithelium leads to spontaneous inflammation and mediates apoptosis in the gut.J Pathol. 2020; 251(2):160-174.