Classifying chronic inflammatory skin diseases by T1-, T2-, T3-, and T4 cytokine signatures for selecting anti-cytokine therapies and restoring healthy skin
Prinicipal Investigator

Dr. Katharina Meier
Dr. Franz Hilke
Scientific interest within the context of the graduate college:
Chronic inflammatory skin diseases with high prevalence such as psoriasis, atopic dermatitis, or lichen planus exemplify how failure of tissue-resident immune regulation can lead to distinct immune response patterns. These skin diseases are manifestations of specific cytokine-driven inflammatory endotypes, commonly referred to as Type 1 (T1; e.g. lichen planus), Type 2 (T2; e.g. atopic dermatitis), Type 3 (T3; e.g. psoriasis), and Type 4 (T4) immune diseases. Type 4 inflammation includes fibrosing (T4a) and granulomatous (T4b) patterns that are relevant for diseases such as morphea and cutaneous sarcoidosis. Understanding the cytokine signaling signatures underlying these diseases offers not only diagnostic precision but also enables rational therapeutic selection. This project integrates cytokine profiling into the framework of skin health and resilience, supporting the overarching goal of the graduate college to define molecular pathways of health, early immune deviation, and preventive intervention.
Project description:
Inflammatory skin diseases are increasingly being classified according to immunological patterns. The classification into T1 (IFN-γ, IL-2, IL-12, TNF), T2 (IL-4, IL-5, IL-13, IL-31), T3 (IL-17A/F, IL-23, IL-1, IL-36), and T4 inflammation (including fibrosing T4a and granulomatous T4b patterns, driven by TGF-β, IL-10, IL-18, and IFN-γ) enables a structured understanding of disease heterogeneity and guides targeted therapy. Currently, more than 25 different anti-cytokine therapeutics are approved for the treatment of immune diseases. Nevertheless, the selection of the bet targeted therapy is indication-based without taking the actual immune signature into account. In this project, we aim to develop and validate a method for characterizing cytokine signatures in inflammatory skin diseases compared to healthy skin. Our goal is not only to improve disease classification but also to enable individualized therapeutic decision-making based on targetable cytokine pathways, thereby supporting the restoration of immune homeostasis. This includes assessing whether treatment-induced cytokine normalization corresponds with clinical remission and molecular markers of tissue recovery. To this end, we will establish a diagnostic framework based on cytokine profiles from retrospective FFPE samples and prospective patient cohorts. The ultimate goal is to identify actionable immune patterns for patient stratification and to better define molecular transitions from health to disease. This approach may help to prevent flares of cytokine-driven skin diseases.
Aim 1: Retrospective cytokine profiling in FFPE skin tissue samples. Patients with newly diagnosed or actively treated chronic inflammatory skin diseases will be recruited for the study. To monitor dynamic changes in tissue inflammation, skin biopsy samples will be obtained at baseline and at defined time points during the course of therapy. In parallel, serum samples will be subjected to multiplex cytokine analysis using high-throughput platforms such as Olink or Luminex to capture systemic immune signatures. The resulting cytokine profiles will be correlated with histological findings and clinical parameters to identify meaningful associations between immune activation patterns, disease characteristics, and treatment response. Based on these data, a cytokine-based scoring index will be developed to classify inflammation types and to support the prediction of therapeutic outcomes.
Aim 2: Prospective validation in serum and fresh skin biopsies. Patients with newly diagnosed or actively treated chronic inflammatory skin diseases will be recruited for the study. Skin biopsy samples will be collected prior to therapy initiation and at defined time points during treatment to capture dynamic changes in tissue inflammation. In parallel, serum samples will be analyzed using multiplex cytokine profiling platforms such as Olink or Luminex to assess systemic immune signatures. The resulting cytokine profiles will be correlated with histological findings and clinical parameters to identify markers associated with disease activity and therapeutic response. Based on these data, a cytokine-based scoring index will be derived to classify inflammation types and support the prediction of treatment outcomes.
References
- Eyerich K and Eyerich S. Immune response patterns in non-communicable inflammatory skin diseases. J Eur Acad Dermatol Venereol. 2018; 32(5):692-703.
- Garzorz-Stark N, Weidinger S, Sticherling M, Ghoreschi K, Enk A, Eyerich K. Inflammatory Skin Diseases: The Importance of Immunological Signatures. Dtsch Arztebl Int. 2025. Online ahead of print.
- Ghoreschi K, Laurence A, Yang XP, Hirahara K, O’Shea JJ. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011; 32(9):395-401.
- Ayres JS. The Biology of Physiological Health. Cell. 2020; 181(2):250-269.
- Gronke K, Hernández PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, […], Glatt H, Triantafyllopoulou A, Diefenbach A. IL-22 protects intestinal stem cells against genotoxic stress. Nature. 2019; 566(7743):249-253.
- Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, […], Linker RA, Alm EJ, Müller DN. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017; 551(7682):585-589.
Resilience against kidney damage
Principal Investigator

Dr. Jan Klocke
Scientific interest within the context of the graduate college:
Our research group focuses on the complex interactions between immune cells and kidney epithelial cells. By analyzing cells found in urine, examining kidney biopsy samples, and utilizing cell culture models, we aim to gain deeper insights into the mechanisms underlying kidney injury and repair. Our work centers on the study of human patient samples, allowing us to address the fundamental question of how kidneys are damaged during disease and how they recover following injury.
Project description:
Kidney damage, in the acute form as acute kidney injury (AKI) or the chronic form as chronic kidney disease (CKD), is among the most frequent forms of organ damage. AKI and CKD both have a tremendous impact and are associated with increased morbidity and mortality, having a large negative impact on global health. In the proposed project we are interested in cellular mechanisms protecting us from kidney injury. Based on our previous observation, immune and kidney tubular epithelial cells can be found in the urine and correlate with damage in AKI and inflammation in CKD.1,2 These cells phenotypically resemble tissue cells, and can be used as a “window into the kidney” to understand pathophysiological processes non-invasively.1,3 Interestingly, increased amounts of immune and tubular epithelial cells can also be observed in critically ill patients without AKI and in stable CKD patients. At present it is unclear, whether these cells just reflect subclinical damage not detected by standard markers, or whether they reflect an active adaptation to kidney stress. Therefore, we will use a combination of state-of-the-art methods such as single-cell RNA sequencing (scRNAseq) and flow cytometry, aiming to understand which active cellular adaptation protects us from kidney damage. All required cohorts and techniques are established in our group.
WP1: Determine the gene expression of kidney tubular epithelial cells in patients without kidney damage. We will use scRNAseq of immune and kidney tubular epithelial cells excreted in the urine of patients without kidney damage. The patients will include critically ill patients without AKI and patients with stable CKD. The respective data will be compared to our existing data sets from patients with active kidney damage (AKI and different forms of other kidney diseases). This WP will yield differences in cell composition and gene expression between active kidney damage and stable kidney function.
WP2: Derive a flow cytometry panel from scRNAseq data. Based on the differences in cell composition and transcriptomes in WP1, we will establish an antibody-based staining panel to detect the respective differences using flow cytometry. This will enable us to analyze a larger cohort of patients in the subsequent WP.
WP3: Active and failed adaptation to kidney stress. We will use flow cytometry and the staining developed in WP2 to assess a larger cohort of patients with kidney stress over time. The cohort will include CKD patients with and without stable kidney function as well as critically ill patients with and without AKI.
References
- Klocke J, Kim SJ, Skopnik CM, Hinze C, Boltengagen A, Metzke D, […], Eckardt KU, Rajewsky N, Enghard P. Urinary single-cell sequencing captures kidney injury and repair processes in human acute kidney injury. Kidney Int. 2022; 102(6):1359-1370.
- Prskalo L, Skopnik CM, Goerlich N, Freund P, Wagner L, Grothgar E, […], Bieringer M, Schreiber A, Enghard P. Urinary CD4+ T cells Predict Renal Relapse in ANCA-Associated Vasculitis: Results of the PRE-FLARED Study. J Am Soc Nephrol. 2024; 35(4):483-494.
- Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, […], Hacohen N, Diamond B, Accelerating Medicines Partnership in SLE Network. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. 2019; 20(7):902-914.
Composition and functional activity of group 2 innate lymphoid cells at the fetal-maternal interface
Prinicipal Investigator
Prof. Dr. Thorsten Braun
Scientific interest within the context of the graduate college:
The goal of my laboratory is to understand the regulation and control mechanisms of immune responses at barrier sites. Barrier sites such as the lungs are continuously exposed to the environment and potential entry ports for pathogens. A unique network of cell populations ensures appropriate immune responses to defend the body against potential pathogens but also to maintain organ function and health at steady state. However, the underlying mechanisms are poorly understood. Group 2 innate lymphoid cells (ILC2) are tissue resident and able to secrete large amounts of cytokines within a short time period and thereby orchestrate innate but also adaptive immune responses. Thus, studying the regulatory processes of ILC2 effector function is key to understand immune concepts of immunity and health at barrier sites.
Project description:
Pregnancy is accompanied by distinct changes to the maternal immune system to provide protection to both the mother and the fetus. Fetal-maternal tissues are unique barrier sites between these two organisms. Immune cells including group 2 innate lymphoid cells (ILC2) are present, however, their contribution to the maintenance of pregnancy as well as the regeneration of tissue is only incompletely understood. Using novel isolation methods in combination with multicolor flow cytometry, we aim to firstly (Aim1) decipher the composition of innate lymphoid cell populations focusing on ILC2 at different locations of the placenta and secondly (Aim2) functionally analyze their activity. Thereby we will gain insights into the mechanisms of ILC2 regulation and function at this unique fetal-maternal site. With our work we further hope to provide an expansion on our knowledge of basic research on innate type 2 immune responses to better understand and potentially promote pathways that contribute to a successful and healthy pregnancy.
References
- Lloyd CM and Marsland BJ. Lung Homeostasis: Infuence of Age, Microbes, and the Immune System. Immunity. 2017; 46(4):549-561.
- Artis D and Spits H. The biology of innate lymphoid cells. Nature. 2015; 517(7534):293-301.
- Saluzzo S, Gorki AD, Rana BMJ, Martins R, Scanlon S, Starkl P, […], Mesteri I, McKenzie AHJ, Knapp S. First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep. 2017; 18(8):1893-1905.
- Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, Bluestone JA, Locksley RM. Interleukin-33 and Interferon-gamma Counter-Regulate Group 2 Innate Lymphoid Cell Activation during Immune Perturbation. Immunity. 2015; 43(1):161-174.
- Duerr CU, McCarthy CDA, Mindt BC, Rubio M, Meli AP, Pothlichet J, […], King IL, Sarfati M, Fritz JH. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. 2016; 17(1):65-75.
- Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, […], Asano K, Betsuyaku T, Koyasu S. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. 2016; 17(1):76-86.
- Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The Microbiome and the Respiratory Tract. Annu Rev Physiol. 2016; 78:481-504.
Dissecting the role of tumor-infiltrating clonal hematopoiesis in patients with gastrointestinal carcinomas
Principal Investigator

Scientific interest within the context of the graduate college:
The scientific focus of our lab centers on unraveling the multifaceted role of clonal hematopoiesis (CH) in disease prevention and inflammatory processes. CH is recognized as a pre-malignant condition that significantly increases the risk of hematologic malignancies. Beyond its oncogenic potential, CH has emerged as a critical risk factor for cardiovascular diseases such as stroke, myocardial infarction, and atherosclerosis, contributing to both initial and recurrent events. Moreover, CH is intricately linked to chronic inflammation, functioning both as a driver and a consequence of sustained immune dysregulation. By investigating these complex interconnections, our research aims to deepen the understanding of CH as a central node in disease pathogenesis and to identify novel strategies for early intervention and prevention.
Project description:
Clonal hematopoiesis (CH), defined by the acquisition of somatic mutations in hematopoietic stem cells, occurs in 20% to 30% of individuals >60 years. CH is associated with a higher overall mortality and an approximately 10-fold risk for the development of hematologic malignancies.1 Reduced overall survival in individuals with CH is mainly caused by an increased rate of cardiovascular events.2 A causal relation was found in preclinical models, showing accelerated development of atherosclerosis driven by an altered function of the NLRP3/IL1β inflammasome of mutated monocytes/macrophages.3 These results pinpoint toward pleiotropic effects of mutated clones, not only affecting self-renewal and differentiation of hematopoietic stem cells but also inflammatory signaling of mature blood cells.
In patients with solid cancer, a very recent report provided evidence that mutated CH-clones can infiltrate solid cancers in up to 30% of patients and remodel the tumor microenvironment and regulate malignant progression. This phenomenon is termed tumor-infiltrating CH (TI-CH).4 While the discovery of TI-CH sets the stage for an entirely new concept of oncogenesis and our understanding for respective tumor cell-microenvironment-interactions, thorough studies in clearly defined patient cohorts – preferentially treated within clinical trials – is warranted to understand how TI-CH may alter response to therapies, cytotoxic side-effects, disease recurrence and patients’ outcome.
The Damm lab has well-documented expertise in investigating CH in large-patient cohorts and has established the entire wet- and dry-lab pipelines to successfully conduct such complex experiments.5,6 In close collaboration with the leading study group AG FIRE (Prof. Modest & Prof. Stintzing, Charité), we will investigate paired tumor and blood samples from more than 750 patients with gastrointestinal cancers. All patients were treated within prospective clinical phase 2 and 3 trials ensuring highest quality of clinical data and direct availability of respective patient specimen.
Aim 1: Defining the prevalence of CH and TI-CH in gastrointestinal cancer. Approximately 750 paired tumor and blood samples from patients with pancreatic cancer (PDAC) and metastatic colorectal cancer (mCRC) treated within prospective clinical phase 2 (PanaMa7) and phase 3 trials (CONKO-58, FIRE-49) will be analyzed for the presence of CH and TI-CH. Error-corrected target sequencing comprising 45 CH-associated genes will be performed using a fully established and automated workflow. CH mutations will be called using our in-house variant calling pipeline as recently published.10
Aim 2: Identification of CH and TI-CH related clinical phenotypes. Next, we will use the obtained genomic results from aim 1 and couple them with patients’ clinical characteristics. To this end, statistical analyses in uni- and multivariable models will be performed using the unique datasets within the clinical trials including primary and secondary study endpoints, but also benefitting from the extensive adjacent translational research projects which will offer discovery of unprecedent (e.g. already existing whole-transcriptome and mutation data, cfDNA).
References
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, [….], Neuberg D, Altshuler D, Ebert BL. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014; 371(26):2488-2498.
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, […], Libby P, Kathiresan S, Ebert BL. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017; 377(2):111-121.
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, […], Hirschi KK, Martin KA, Walsh K. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017; 355(6327):842-847.
- Pich O, Bernard E, Zagorulya M, Rowan A, Pospori C, Slama R, […], Bonnet D, Papaemmanuil E, Swanton C . Tumor-Infiltrating Clonal Hematopoiesis. N Engl J Med. 2025; 392(16):1594-1608.
- Arends CM, Dimitriou S, Stahler A, Hablesreiter R, Strzelecka PM, Stein CM, […], Stintzing S, Heinemann V, Damm F. Clonal hematopoiesis is associated with improved survival in patients with metastatic colorectal cancer from the FIRE-3 trial. Blood. 2022; 139(10):1593-1597.
- Arends CM, Liman TG, Strzelecka PM, Kufner A, Löwe P, Huo S, […], Weber JE, Endres M, Damm F. Associations of clonal hematopoiesis with recurrent vascular events and death in patients with incident ischemic stroke. Blood. 2023; 141(7):787-799.
- Modest DP, Karthaus M, Fruehauf S, Graeven U, Müller L, König AO, […], Heinemann V, Stintzing S, Trarbach T. Panitumumab Plus Fluorouracil and Folinic Acid Versus Fluorouracil and Folinic Acid Alone as Maintenance Therapy in RAS Wild-Type Metastatic Colorectal Cancer: The Randomized PANAMA Trial (AIO KRK 0212). J Clin Oncol. 2022; 40(1):72-82.
- Hoyer K, Hablesreiter R, Inoue Y, Yoshida K, Briest F, Christen F, […], Ogawa S, Sinn M, Damm F. A genetically defined signature of responsiveness to erlotinib in early-stage pancreatic cancer patients: Results from the CONKO-005 trial. EBioMedicine. 2021; 66:103327.
- Stintzing S, Klein-Scory S, Fischer von Weikersthal L, Fuchs M, Kaiser F, Heinrich K, […], Schmiegel W, Baraniskin A, Heinemann V. Baseline Liquid Biopsy in Relation to Tissue-Based Parameters in Metastatic Colorectal Cancer: Results From the Randomized FIRE-4 (AIO-KRK-0114) Study. J Clin Oncol. 2025; 43(12):1463-1473.
- Arends CM, Kopp K, Hablesreiter R, Estrada N, Christen F, Moll UM, […], Bullinger L, Braicu EI, Damm F. Dynamics of clonal hematopoiesis under DNA-damaging treatment in patients with ovarian cancer. Leukemia. 2024; 38(6):1378-1389.
Role of extracellular sulfate on vascular health
Principal Investigator

Scientific interest within the context of the graduate college:
Sulfate is an essential nutrient that supports numerous biological processes, including tissue development, cellular function, and overall physiological homeostasis. Its concentration in the body is tightly regulated by specialized sulfate transporters that mediate both dietary absorption and renal reabsorption. Despite its fundamental roles, the contribution of sulfate to vascular biology has been largely overlooked. Emerging evidence suggests that sulfate is critical for maintaining the structural and functional integrity of blood vessels, particularly through its incorporation into sulfated glycosaminoglycans, which are key components of the endothelial glycocalyx. We hypothesize that sulfate plays a vital role in preserving vascular integrity, supporting endothelial function, and regulating blood flow-domains that remain underexplored and represent significant knowledge gaps in biomedical research.
Project description:
We recently identified a subset of patients with impaired capacity to maintain normal blood sulfate concentrations due to damaging variants in the sulfate transporter geneSLC26A1.1 These individuals present with musculoskeletal disorders, which we suspect are linked to systemic sulfate deficiency.2 To investigate the underlying mechanisms, we developed a novel mouse model with tissue-specific deletion ofSLC26A1using Cre-Lox technology. Our findings indicate that global and kidney-specific deletion ofSLC26A1leads to significantly reduced plasma sulfate levels, primarily due to excessive urinary excretion. These mice also exhibit a skeletal phenotype characterized by impaired bone mineralization. To date, the vascular system has not been evaluated in this model. However, preliminary human data show that individuals carrying damagingSLC26A1variants exhibit increased cardiovascular mortality compared to matched controls, suggesting a potential vascular consequence of sulfate deficiency. This project seeks to fill this critical knowledge gap by exploring the relationship between extracellular sulfate levels and vascular health using complementary in vitro, in vivo, and population-based approaches.
Aim 1: Investigate the effects of low extracellular sulfate on vascular cell function in vitro. This aim will assess the impact of sulfate deficiency on endothelial and vascular smooth muscle cells, focusing on key parameters such as cell viability, nitric oxide signaling, glycocalyx integrity, and inflammatory response under defined culture conditions.
Aim 2: Evaluate vascular function in vivo in mice with deletion of SLC26A1. This aim will characterize vascular phenotypes in SLC26A1-deficient mice using histological, biochemical, and physiological assessments to determine whether systemic sulfate deficiency disrupts vascular structure and function.
Aim 3: Examine the association between damaging SLC26A1 variants and increased cardiovascular mortality in the Mayo Clinic Biobank. Leveraging data from the Mayo Clinic Biobank and the Tapestry Study, our current Re-Thinking Health student Felix Pitzken has identified that approximately 3% of over 150,000 individuals carry potentially damaging variants in SLC26A1. With support from our biostatistics team, this aim will evaluate whether these variants are associated with increased cardiovascular risk, and whether this risk is mediated by reduced plasma sulfate concentrations.
References
- Pfau A, López-Cayuqueo KI, Scherer N, Wuttke M, Wernstedt A, González Fassrainer D, […], Köttgen A, Jentsch TJ, Knauf F. SLC26A1 is a major determinant of sulfate homeostasis in humans. J Clin Invest. 2023; 133(3):e161849.
- Scherer N, Fässler D, Borisov O, Cheng Y, Schlosser P, Wuttke M, […], Thiele I, Hertel J, Köttgen A. Coupling metabolomics and exome sequencing reveals graded effects of rare damaging heterozygous variants on gene function and human traits. Nat Genet. 2025; 57(1):193-205.
Senescent cells in progression of IBD to CRC
Principal Investigator

Scientific interest within the context of the graduate college:
Our group aims to understand the molecular mechanisms involved in tissue homeostasis, inflammation, and resolution of inflammation. Our main focus is on the transcription factor NF-κB and its role in the intestinal epithelium. Our current projects range from determining the role of the transcription factor in epithelial regeneration in colitis and in inflammatory bowel diseases (Re-Thinking Health, 2022), to refining analgesia (Charité 3R), to investigating the role of NF-κB in cellular senescence in the gut (DFG) or its role in metabolism.
Project description:
Senescent cells are characterized by terminal cell cycle arrest, epigenetic changes, and by the senescence associated secretory phenotype (SASP). SASP is comprised of scores of cytokines, chemokines, and growth factors, which regulate cells and tissues in a paracrine manner. Therefore, even a small number of senescent cells can have a dramatic impact on distant tissues and were shown to sustain inflammation, fuel carcinogenesis, and shorten lifespan. Most of the genes that code for SASP factors are transcriptionally regulated by NF-κB8.1,2 Under physiological conditions SASP promotes wound healing and recruitment of immune cells, which help clear senescent cells.
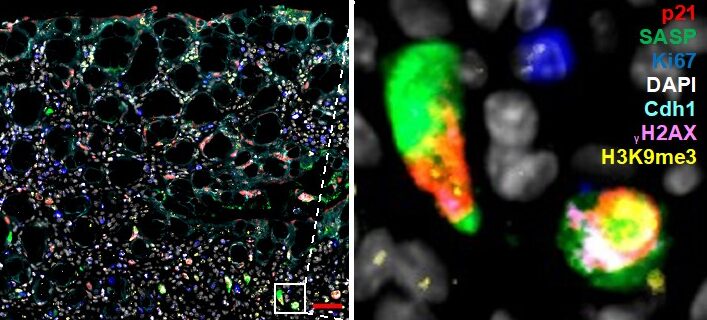
Figure 1. Multiplex immunofluorescence staining of senescence. Mouse colon (colitis model) was stained with markers for senescence. Simultaneous staining with numerous markers (multiplex) is required to rule out other cell states (proliferation, apoptosis, quiescence). Zoom-in on select fully senescent cells is shown on the right.
We are interested in investigating what kind of senescent populations arise in inflammatory bowel diseases (IBD) versus those that progressed to colorectal cancer (CRC). We are especially interested in the role that SASP plays in disease progression and whether distinct forms of SASP can be exploited therapeutically.
As part of our preliminary data, we have already identified distinct populations dependent on stage of inflammation and importantly, we show that no single type of senescence exists, but rather cells display different proficiencies for SASP and thus for communication with their environment. It is not known how these differences skew progression of IBD.
Aim 1: Characterize senescent cells in IBD and CRC and in murine colitis and CRC models. As part of this work package you will perform multiplex stainings (as in Figure 1) using already available samples from IBD and CRC patient samples and from murine mouse models. You will also receive training in basic analysis of single-cell RNA sequencing and will therefore quantify cells that have SASP versus those that do not. You will then isolate these cells using FACS and determine how different types of SASP alter immune cell recruitment.
Aim 2: Reprogram senescent cells to transform immunosuppressive “cold” tumor microenvironment environment into “hot”. Here you will use available senolytics and senomorphics (drugs that kill senescent cells or suppress their SASP), and those identified via our collaborators through deep learning, to reprogram senescent cells in 2D culture and in human and mouse intestinal organoids. Successful senolytics will be used in pre-clinical trials in CRC mouse models to determine if these expose tumors to immunosurveillance.
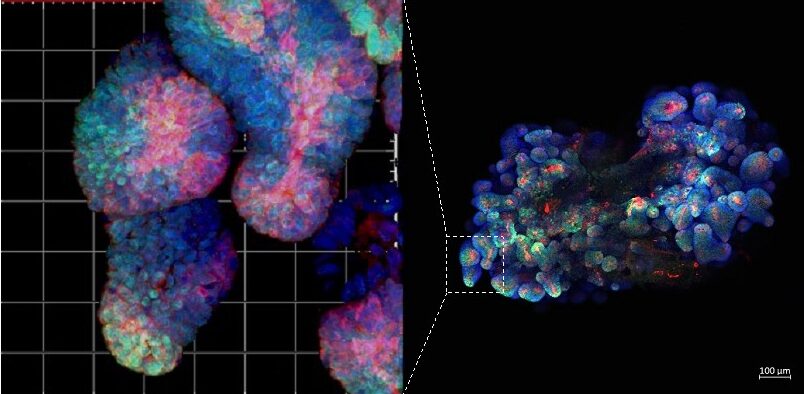
Figure 2. Immunofluorescence staining of mouse fluorescent organoid. All our projects combine work with mouse models. patient samples (including organoids) and bioinformatics.
Methods you will learn as part of this project: multiplex immunofluorescent staining, single-cell data analysis, immunohistochemistry, human and mouse organoid cultures (Figure 2), qPCR.
References
- Kolesnichenko M, Mikuda N, Höpken UE, Kärgel E, Uyar B, Tufan AB, […], Schmidt-Ullrich R, Schmitt CA, Scheidereit C. Transcriptional repression of NFKBIA triggers constitutive IKK- and proteasome-independent p65/RelA activation in senescence. EMBO J. 2021; 40(6):e104296.
- Mikuda N, Schmidt-Ullrich R, Kärgel E, Golusda L, Wolf J, Höpken UE, Scheidereit C, Kühl AA, Kolesnichenko M. Deficiency in IκBα in the intestinal epithelium leads to spontaneous inflammation and mediates apoptosis in the gut. J Pathol. 2020; 251(2):160-174.