Unraveling the role of a macrophage-B cell loop in the immune microenvironment in chronic liver disease using spatial proteomics and intravital imaging
Prinicipal Investigator

Dr. Felix Heymann
Scientific interest within the context of the graduate college
Chronic liver disease is among the most prevalent causes of organ failure and death worldwide, yet the molecular and cellular mechanisms that drive its progression remain incompletely understood and are driven by different etiologies. Metabolic dysfunction-associated steatotic liver disease (MASLD) and alcoholic liver disease (ALD) account for the majority of cases with liver cirrhosis in Germany. Our group investigates how the hepatic immune microenvironment is reshaped during chronic inflammation, with a particular focus on the spatial organization of innate immune cells. Kupffer cells and monocyte-derived macrophages occupy defined tissue niches within the liver, and their function is strongly influenced by the local molecular environment. Similarly, B cells accumulate in diseased liver tissue and produce immunoglobulin, however this relationship in the pathogenesis of chronic liver disease remains unexplored. This project sits at the heart of the Re-Thinking Health mission: by mapping the spatial protein landscape of the inflamed and fibrotic liver and linking it to the real-time behavior of immune cells, we aim to define the molecular hallmarks that distinguish healthy from diseased hepatic tissue neighborhoods. The findings will provide a foundation for understanding how immune dysregulation is spatially organized – and how it might be interrupted to preserve or restore liver health.
Project description
Liver cirrhosis is the shared endpoint of most chronic liver diseases and is characterized by progressive fibrosis, sustained immune infiltration, and a fundamental remodeling of the hepatic tissue architecture. Despite its clinical importance, the spatial protein landscape of the fibrotic liver and the molecular neighborhoods occupied by innate immune cells has not been systematically characterized. Our group has generated preliminary data using laser-microdissection followed by protein mass spectrometry, revealing striking enrichment of immunoglobulin subunits and complement proteins (including C1q, C3, and C3b) in fibrotic and inflammatory lesion areas. These findings suggest that B cell-derived antibodies and the downstream complement cascade are spatially concentrated in regions of active liver damage, where they likely amplify macrophage activation and perpetuate tissue injury. This project will characterize these processes from two complementary angles: first, by generating a spatially resolved protein atlas of the fibrotic liver using MALDI imaging mass spectrometry; and second, by observing the behavior of macrophages and B cells in the liver by intravital multiphoton microscopy, with particular attention to complement-dependent interactions.
Aim 1: Spatial proteomics atlas of the complement-immune landscape in liver cirrhosis. We will begin by analyzing an existing dataset of spatially resolved protein mass spectrometry data generated from murine liver tissue across key disease stages: healthy controls, chronic inflammation with fibrosis, and disease regression. This analysis will identify proteins and protein networks enriched in specific tissue compartments – including periportal infiltrates, fibrotic septa, and sinusoidal regions – with a particular focus on immunoglobulins, complement components (C1q, C3, C3b, C5), and complement receptors expressed on macrophages (CD11b, CD16, CRIg). Building on these findings, we will perform MALDI imaging mass spectrometry on tissue sections from patients with liver cirrhosis and from mice after chronic CCl4-induced liver injury. MALDI imaging will provide high-resolution spatial maps of protein and lipid distributions across intact tissue sections without the need for prior region selection, allowing an unbiased comparison of molecular neighborhoods between species and disease stages. Together, the two approaches will generate a spatially resolved atlas linking immunoglobulin deposition and complement activation to defined inflammatory microenvironments in the diseased liver..
Aim 2: Intravital imaging of B cell and macrophage dynamics in the context of complement activation. The spatial protein maps generated in Aim 1 will be complemented by real-time imaging of immune cell behavior in the living liver. Using multiphoton intravital microscopy in fluorescent reporter mice together with antibody labeling, we will characterize how macrophages and B cells localize within the tissue, interact with one another, and respond to the local complement-rich environment. Complement receptor-dependent macrophage activation will be assessed by measuring phagocytic uptake of C3b-opsonized fluorescent particles in vivo, allowing direct comparison between healthy and chronically injured livers. B cell–macrophage contact events will be quantified by measuring interaction frequency and duration. These experiments will be performed in healthy controls and at defined stages of CCl4-induced liver injury, including active inflammation and the regression phase. By anchoring the dynamic cellular observations to the spatial proteomic maps from Aim 1, the project will reveal an integrated map of how complement deposition organizes the macrophage–B cell inflammatory axis in different stages of chronic liver disease.
References
- Peiseler M, Araujo David B, Zindel J, Surewaard BGJ, Lee WY, Heymann F, Nusse Y, Castanheira FVS, Shim R, Guillot A, Bruneau A, Atif J, Perciani C, Ohland C, Ganguli Mukherjee P, Niehrs A, Thuenauer R, Altfeld M, Amrein M, Liu Z, Gordon PMK, McCoy K, Deniset J, MacParland S, Ginhoux F, Tacke F, Kubes P. Kupffer cell-like syncytia replenish resident macrophage function in the fibrotic liver. Science. 2023; 381(6662): eabq5202.
- Scholten D, Trebicka J, Liedtke C, Weiskirchen R. The carbon tetrachloride model in mice. Lab Anim. 2015; 49(1 Suppl): 4-11.
- Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017; 66(6): 1300-1312.
- Guidotti LG, Inverso D, Sironi L, Di Lucia P, Fioravanti J, Ganzer L, Fiocchi A, Vacca M, Aiolfi R, Sammicheli S, Mainetti M, Cataudella T, Raimondi A, Gonzalez-Aseguinolaza G, Protzer U, Ruggeri ZM, Chisari FV, Isogawa M, Sitia G, Iannacone M. Immunosurveillance of the liver by intravascular effector CD8+ T cells. Cell. 2015; 161(3): 486–500.
- Heymann F, Tacke F. Immunology in the liver – from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016; 13(2): 88-110.
- Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwälder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease – novel insights into cellular communication circuits. J Hepatol. 2022; 77(4): 1136-1160.
- Wang Y, Heymann F, Peiseler M. Intravital imaging: dynamic insights into liver immunity in health and disease. Gut. 2024; 73(8): 1364–1375.
- Oleinika K, Mauri C, Salama AD. Effector and regulatory B cells in immune-mediated kidney disease. Nat Rev Nephrol. 2019; 15(10): 622-638.
- Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009; 30(2): 83-90.
- Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, Taylor RS, Efremova M, Vento-Tormo R, Carragher NO, Kendall TJ, Fallowfield JA, Harrison EM, Mole DJ, Wigmore SJ, Newsome PN, Weston CJ, Iredale JP, Tacke F, Pollard JW, Ponting CP, Marioni JC, Teichmann SA, Henderson NC. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019; 575(7783): 512–518.
Ultra-sensitive systemic inflammatory signatures of age-related multimorbidity in naturally aged mice
Prinicipal Investigator

Scientific interest within the context of the graduate college
This project addresses a central aim of the EKFS Graduate School: to identify systemic signals that precede, accompany, and potentially predict age-related disease. Rather than studying one organ in isolation, the unbiased ageing-health project asks whether a measurable low-grade inflammatory state in blood marks broader multimorbidity in ageing. By linking ultra-sensitive circulating inflammatory profiles to visible, metabolic, neoplastic, ocular, and microbiome-associated ageing phenotypes in mice, the study fits the prevention focus of EKFS and can nominate biomarker signatures relevant to early risk stratification in humans.
Project description
Chronic low-grade inflammation (“inflammaging”) is increasingly viewed as a shared upstream process across many late-life disorders, yet it remains unclear whether one measurable systemic inflammatory state tracks the breadth of spontaneous age-related pathology within an individual. Naturally aged C57BL/6J mice provide a useful discovery model because genetically identical animals nevertheless develop heterogeneous phenotypes including neoplasia, metabolic dysfunction and hepatic steatosis, coat deterioration with alopecia and hair greying, cataract, and marked shifts in the gut microbiome. This EKFS proposal defines a smaller and more feasible student subproject from a large unbiased murine ageing study run in the Latz lab centered on one tractable question: are ultra-sensitive blood inflammatory signatures linked to these age-related phenotypes? Using the Alamar Biosciences NULISAseq Mouse Panel 120, a murine multiplex assay with 120+ protein targets, attomolar sensitivity, and low-input workflows, the student will analyze archived serum or plasma from an existing aged mouse cohort and integrate these data with available pathology and microbiome readouts. The overall goal is to derive a simple systemic inflammaging score and test whether it identifies mice with a higher burden of spontaneous age-related disease.
Aim 1 / WP1: Generate ultra-sensitive systemic inflammatory profiles in naturally aged mice. Archived serum or plasma from naturally aged C57BL/6J mice will be analyzed using the Alamar Biosciences NULISAseq Mouse Panel 120. This work package is intentionally focused and technically feasible for a medical student because it relies on existing samples and an established multiplex platform rather than de novo animal experimentation. After standard quality control and normalization, the student will quantify circulating cytokines, chemokines, and related inflammatory mediators and construct a compact inflammaging score based on the most reproducible markers. In addition to describing the overall inflammatory landscape of the cohort, the analysis will identify individual proteins or small marker combinations that segregate mice with low versus high age-related disease burden.
Aim 2 / WP2: Test whether systemic inflammatory signatures are linked to age-related pathologies and microbiome states. The inflammatory profiles generated in Aim 1 will be integrated with existing phenotypic readouts from the same animals. Priority phenotypes will include neoplastic lesions, metabolic syndrome-like traits (for example body weight, glucose dysregulation and/or steatosis where available), coat changes such as alopecia and hair greying, cataract, and microbiome features derived from fecal profiling. The student will establish a multimorbidity index and determine whether the systemic inflammaging score, or selected individual proteins, associates with single phenotypes and with cumulative disease burden across organs. This analysis will provide an experimentally manageable first step toward identifying blood-based markers that report organismal ageing trajectories and will nominate high-priority biomarker-pathology links for subsequent mechanistic follow-up in the larger parent project.
References
- Dugan B, Conway J, Duggal NA. Inflammaging as a target for healthy ageing. Age Ageing. 2023; 52(2): afac328.
- Fulop T, Larbi A, Pawelec G, Khalil A, Cohen AA, Hirokawa K, Witkowski JM, Franceschi C. Immunology of aging: the birth of inflammaging. Clin Rev Allergy Immunol. 2023; 64(2): 109-122.
- Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, Schertzer JD, Larché MJ, Davidson DJ, Verdú EF, Surette MG, Bowdish DME. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. 2017; 21(1): 455-466.
- Feng W, Beer JC, Hao Q, Ariyapala IS, Sahajan A, Komarov A, Cha K, Moua M, Qiu X, Xu X, Iyengar S, Yoshimura T, Nagaraj R, Wang L, Yu M, Engel K, Zhen L, Xue W, Lee CJ, Park CH, Peng C, Zhang K, Grzybowski A, Hahm J, Schmidt SV, Odainic A, Spitzer J, Buddika K, Kuo D, Fang L, Zhang B, Chen S, Latz E, Yin Y, Luo Y, Ma XJ. NULISA: a proteomic liquid biopsy platform with attomolar sensitivity and high multiplexing. Nat Commun. 2023; 14(1): 7238.
- Schaum N, Lehallier B, Hahn O, Pálovics R, Hosseinzadeh S, Lee SE, Sit R, Lee DP, Losada PM, Zardeneta ME, Fehlmann T, Webber JT, McGeever A, Calcuttawala K, Zhang H, Berdnik D, Mathur V, Tan W, Zee A, Tan M; Tabula Muris Consortium; Pisco AO, Karkanias J, Neff NF, Keller A, Darmanis S, Quake SR, Wyss-Coray T. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. 2020; 583(7817): 596-602.
Development of an ECM-based advanced in vitro model to elucidate mechanisms of MASLD onset, recovery, and prevention
Prinicipal Investigator

Dr. Pavitra Kumar
Scientific interest within the context of the graduate college
***Follows.
Project description
Metabolic dysfunction-associated steatotic liver disease (MASLD) is the most prevalent chronic liver disease worldwide and represents a major unmet clinical need. While disease progression, from steatosis to steatohepatitis, fibrosis, and cirrhosis, is well described, the mechanisms governing early disease onset, reversibility, and prevention remain poorly understood.1,2
Emerging evidence highlights the extracellular matrix (ECM) as a dynamic regulator not only of disease progression but also of cellular plasticity, tissue repair, and resolution of inflammation. Changes in ECM composition and stiffness influence hepatocyte metabolism, stellate cell activation, and immune signaling, suggesting that ECM cues may critically determine whether liver tissue progresses toward disease or returns to homeostasis.3
Current in vitro models inadequately capture these ECM-driven processes and largely focus on late-stage disease phenotypes, limiting their utility for identifying early drivers of disease onset or targets for preventive intervention.4 The ECM is increasingly recognized as a key regulator of disease progression by influencing hepatocyte metabolism, stellate cell activation, and inflammatory signaling.5
We hypothesize that ECM derived from livers at distinct stages of MASLD encodes stage-specific biochemical and mechanical signals that actively regulate transitions between healthy, diseased, and recovery states. By incorporating this ECM into a 3D multicellular system, we aim to establish an advanced in vitro model that enables the dissection of mechanisms underlying disease initiation, progression, and reversal, ultimately identifying pathways that can be targeted for prevention. The overall objective of this project is to develop a physiologically relevant ECM-based in vitro platform that integrates disease-stage–specific matrix cues with multicellular liver architecture to investigate mechanisms of MASLD onset, recovery, and prevention.
Aim 1. Isolate and characterize ECM from livers across progressive and early-stage MASLD to define matrix signatures associated with disease onset and progression.
Aim 2. Utilize the ECM-based model to identify and modulate pathways involved in disease reversal and prevention, including testing recovery-inducing conditions and anti-fibrotic/anti-inflammatory interventions.
This project will provide a novel platform to move beyond descriptive models of MASLD toward a mechanistic understanding of disease dynamics, enabling identification of targets for early intervention and prevention strategies. By integrating ECM biology with multicellular modelling, the system has the potential to accelerate the development of therapies aimed at halting or reversing MASLD before irreversible liver damage occurs.
References
- Huang M, Chen H, Wang H, Zhang Y, Li L, Lan Y, Ma L. Global burden and risk factors of MASLD: trends from 1990 to 2021 and predictions to 2030. Intern Emerg Med. 2025; 20(4): 1013-1024.
- Engelmann C, Tacke F. The potential role of cellular senescence in non-alcoholic fatty liver disease. Int J Mol Sci. 2022; 23(2): 652.
- Wu Y, Cao Y, Xu K, Zhu Y, Qiao Y, Wu Y, Chen J, Li C, Zeng R, Ge G. Dynamically remodeled hepatic extracellular matrix predicts prognosis of early-stage cirrhosis. Cell Death Dis. 2021; 12(2): 163.
- Ros-Tarraga P, Villanueva-Badenas E, Sanchez-Gonzalez E, Gallego-Ferrer G, Donato MT, Tolosa L. Challenges of in vitro modelling of liver fibrosis. Front Cell Dev Biol. 2025; 13: 1567916.
- Arteel GE. Hepatic extracellular matrix and its role in the regulation of liver phenotype. Semin Liver Dis. 2024; 44(3): 343-355.
- Kumar P, Hassan M, Tacke F, Engelmann C. Delineating the heterogeneity of senescence-induced-functional alterations in hepatocytes. Cell Mol Life Sci 2024; 81(1): 200.
- Mazza G, Rombouts K, Rennie Hall A, Urbani L, Vinh Luong T, Al-Akkad W, Longato L, Brown D, Maghsoudlou P, Dhillon AP, Fuller B, Davidson B, Moore K, Dhar D, De Coppi P, Malago M, Pinzani M. Decellularized human liver as a natural 3D-scaffold for liver bioengineering and transplantation. Sci Rep. 2015; 5: 13079.
Targeting alveolar macrophage self-renewal to maintain lung homeostasis and prevent chronic inflammatory lung disease
Principal Investigator

Scientific interest within the context of the graduate college
Active maintenance of tissue health requires maintenance of tissue-resident macrophages that perform homeostatic functions. Loss of tissue-resident macrophages reduces the ability of tissues to maintain homeostasis or to return to homeostasis after inflammation. We propose that exploring the mechanisms that maintain tissue-resident macrophages will allow us to identify molecular targets that promote tissue health and to achieve deeper remission after treatment of chronic inflammatory diseases.
Project description
Alveolar macrophages play an active role in maintaining lung homeostasis by clearing apoptotic cells, catabolizing surfactant produced by alveolar epithelial cells and mediating antimicrobial immunity. Loss of alveolar macrophage function results in inability to maintain lung homeostasis, susceptibility to infection and exacerbated inflammatory responses. Despite their importance, the mechanisms that actively maintain alveolar macrophages and thus secure lung homeostasis are largely unknown.
The project will explore the mechanisms that maintain alveolar macrophages. To do this, we will employ transgenic mouse models that allow targeting alveolar macrophages in two different stages of their development. Alveolar macrophages will be isolated using bronchoalveolar lavage and their proliferation capacity, transcriptional program, metabolism and ability to clear apoptotic cells will be studied ex vivo.
Overall, this line of work aims to uncover essential pathways that promote alveolar macrophage maintenance and may be targeted to prevent lung injury and pulmonary fibrosis in patients with autoimmunity.
Role of the sulfate transporter SLC26A1 in protection against acetaminophen (Tylenol, Paracetamol) toxicity
Principal Investigator

, Prof. Dr. Peter Aronson
Scientific interest within the context of the graduate college
Within the graduate college, this project addresses how systemic sulfate homeostasis influences drug-induced liver injury. By studying the role of SLC26A1 in acetaminophen toxicity, it links renal and hepatic physiology with translational and population-based approaches. The integration of mouse models and human biobank data aligns with the program’s goal of bridging basic and clinical research to identify mechanisms and risk factors of acute liver failure.
Project description
Acetaminophen (Tylenol/Paracetamol) toxicity is the leading cause of acute liver failure (ALF) in Western countries. In North America alone, acetaminophen overdose accounts for approximately 50,000 emergency department visits and 500 deaths annually.1 At therapeutic doses, acetaminophen is primarily metabolized in the liver through glucuronidation and sulfation, resulting in nontoxic conjugates that are efficiently excreted by the kidney.2 Sulfation requires the sulfate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS), whose availability is critically dependent on adequate inorganic sulfate levels.3 SLC26A1 is a major sulfate transporter that our group has studied extensively in both murine models and human populations.4-6 Prior studies in mice have demonstrated that loss of SLC26A1 function is associated with increased acetaminophen-induced liver injury and elevated transaminase levels.7 Although SLC26A1 is highly expressed in the liver, it is also abundantly present in the intestine and kidney. A key unresolved question is whether heightened acetaminophen toxicity associated with impaired SLC26A1 function is driven by reduced systemic sulfate availability (due to renal sulfate wasting) or by impaired local sulfate transport within hepatocytes. We hypothesize that increased acetaminophen toxicity in SLC26A1 deficiency is primarily attributable to reduced plasma sulfate levels resulting from renal sulfate loss, rather than impaired sulfate transport within the liver itself. To test this hypothesis, we will induce acetaminophen-mediated liver injury in mice using an established protocol and compare outcomes across wild-type mice, whole-body SLC26A1 knockout mice, and tissue-specific SLC26A1 knockout models targeting the kidney or liver. In parallel, we have identified approximately 1,400 individuals with damaging SLC26A1 variants within the Mayo Clinic Biobank. We will assess baseline liver function, specifically serum transaminase levels, in these individuals compared with matched controls. Additionally, we will perform Mendelian randomization analyses to determine whether genetic variants in SLC26A1 are associated with higher transaminase levels in the context of acetaminophen exposure. In summary, this proposed doctoral thesis aims to address a critical knowledge gap by integrating mechanistic studies in genetically engineered mouse models, already generated and available, with large-scale, population-based analyses at the Mayo Clinic.
Aim 1: Determine the impact of tissue-specific deletion of SLC26A1 on acetaminophen-induced liver toxicity. This aim will compare acetaminophen toxicity in whole-body SLC26A1 knockout mice versus mice with kidney-and liver-specific deletion of SLC26A1, thereby isolating the contribution of renal sulfate handling.
Aim 2:Evaluate the association between damaging SLC26A1 variants, liver injury markers, and acetaminophen exposure in humans. Using data from the Mayo Clinic Biobank and the Tapestry Study, we will examine whether individuals harboring damaging SLC26A1 variants exhibit higher transaminase levels compared with matched controls, and whether acetaminophen intake further amplifies this association.
References
- Fontana RJ, Liou I, Reuben A, Suzuki A, Fiel MI, Lee W, Navarro V. AASLD practice guidance on drug, herbal, and dietary supplement-induced liver injury. Hepatology. 2023; 77(3): 1036-1065.
- McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013; 30(9): 2174-2187.
- Klaassen CD, Boles JW. Sulfation and sulfotransferases 5: the importance of 3′-phosphoadenosine 5′-phosphosulfate (PAPS) in the regulation of sulfation. FASEB J. 1997; 11(6): 404-418.
- Pfau A, López-Cayuqueo KI, Scherer N, Wuttke M, Wernstedt A, González Fassrainer D, Smith DE, van de Kamp JM, Ziegeler K, Eckardt KU, Luft FC, Aronson PS, Köttgen A, Jentsch TJ, Knauf F. SLC26A1 is a major determinant of sulfate homeostasis in humans. J Clin Invest. 2023; 133(3): e161849.
- Scherer N, Fässler D, Borisov O, Cheng Y, Schlosser P, Wuttke M, Haug S, Li Y, Telkämper F, Patil S, Meiselbach H, Wong C, Berger U, Sekula P, Hoppmann A, Schultheiss UT, Mozaffari S, Xi Y, Graham R, Schmidts M, Köttgen M, Oefner PJ, Knauf F, Eckardt KU, Grünert SC, Estrada K, Thiele I, Hertel J, Köttgen A. Coupling metabolomics and exome sequencing reveals graded effects of rare damaging heterozygous variants on gene function and human traits. Nat Genet. 2025; 57(1): 193-205.
- Ko N, Knauf F, Jiang Z, Markovich D, Aronson PS. Sat1 is dispensable for active oxalate secretion in mouse duodenum. Am J Physiol Cell Physiol. 2012; 303(1): C52-57.
- Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, Clarke LA, Markovich D. Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J Clin Invest. 2010; 120(3): 706-712.
Group 3 innate lymphoid cell regulation of vascular remodeling at homeostasis and in cancer
Principle Investigator

Scientific interest within the context of the graduate college
We are interested in understanding the mechanisms by which a specific subset of innate lymphocytes, namely group 3 innate lymphoid cells (ILC3), regulate immune responses and tissue functions at homeostasis and in cancer. ILC3 are tissue-resident cells located at barrier tissues which occupy specific tissue niches where they constantly produce cytokines, particularly IL-22. ILC3-derived IL-22 maintains the integrity of epithelial surfaces and regulates cell processes involved in epithelial cell malignant transformation. However, the role of ILC3 and IL-22 in vascular remodeling and angiogenesis, which is a key step in tumorigenesis and a hallmark of cancer, remains largely unexplored. We seek to systematically investigate the role of ILC3 in the regulation of endothelial cell development and function in homeostasis and during tumor evolution to identify new therapeutic targets.
Project description
ILC3 are tissue-resident IL-22-producing cells that play an important role in the adaptation of tissues to environmental changes and stressors.1 IL-22 is a cytokine belonging to the IL-10 family that exerts regulatory functions by binding to its receptor IL-22RA1, selectively expressed by non-hematopoietic cells.2 Unpublished work from our laboratory recently showed that IL-22 regulates melanoma angiogenesis by inducing molecular programs in tumor endothelial cells that promote the formation of aberrant, dysfunctional and leaky tumor vessels. This argues for potential ILC3-endothelial cell pathways regulating the formation of new vessels at barrier tissues that can be co-opted by the tumor “neo-organ”. Blood vessels are crucial to gut function and pervade all intestinal tissue layers.3 The gut vascular barrier (GVB) is composed of closely interacting endothelial cells that display unique features.3,4 Alteration of the GVB due to pathological tissue remodeling leads to the leakage of the gut and the development of intestinal and extraintestinal diseases.2,4 A better understanding of the ILC3-dependent regulation of endothelial cell differentiation and of their function in the intestine could lead to the identification of an ILC3-endothelial cell module regulating intestinal homeostasis and tumor progression, promoting the development of novel therapeutic approaches to treat cancer.5,6
Aim 1: To investigate ILC3 regulation of intestinal endothelial cell development and function. The small intestine is a very plastic organ which undergoes postnatal dynamic changes.7 Our preliminary results suggest that IL-22 has a role in the regulation of physiological postnatal vascular remodeling. Genetically modified mouse models will be used to explore the molecular mechanisms by which ILC3 and IL-22 contribute to intestinal vasculogenesis from early to adult mouse life. Molecular (qPCR, RNA-sequencing) and cell biology (flow cytometry, immunofluorescence) approaches will be employed. In vivo functional vascular assays will be also applied.
Aim 2: To investigate ILC3 regulation of angiogenesis in intestinal cancer. After the identification of ILC3 and IL-22-dependent pathways regulating physiological vascular remodeling, we will establish preclinical models of intestinal cancer, either inflammation-induced or oncogene-induced. Mouse surgery and histopathological analysis, as well as molecular and cellular biology techniques will be used to investigate the role of the ILC3-endothelial cell module in cancer onset and progression.
References
- Mattiola I, Diefenbach A. Innate lymphoid cells and cancer at border surfaces with the environment. Semin Immunol. 2019; 41: 101278.
- Mattiola I, Diefenbach A. Regulation of innate immune system function by the microbiome: consequences for tumor immunity and cancer immunotherapy. Semin Immunol. 2023; 66: 101724.
- Bernier-Latmani J, González-Loyola A, Petrova TV. Mechanisms and functions of intestinal vascular specialization. J Exp Med. 2024; 221(1): e20222008.
- Brescia P, Rescigno M. The gut vascular barrier: a new player in the gut-liver-brain axis. Trends Mol Med. 2021; 27(9): 844-855.
- Ebeling S, Kowalczyk A, Perez-Vazquez D, Mattiola I. Regulation of tumor angiogenesis by the crosstalk between innate immunity and endothelial cells. Front Oncol. 2023; 13: 1171794.
- Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011; 17(11): 1359-1370.
- Rakoff-Nahoum S, Kong Y, Kleinstein SH, Subramanian S, Ahern PP, Gordon JI, Medzhitov R. Analysis of gene-environment interactions in postnatal development of the mammalian intestine. Proc Natl Acad Sci U S A. 2015; 112(7): 1929-1936.
The nuclear import and stability of GATA-3 as a central rheostat for lymphocyte identity OR Elucidating the structural basis and functional relevance of GATA-3 foci in lymphocytes
Principle Investigator

Dr. Philippe Saikali
Scientific interest within the context of the graduate college
PROJECT 1. The nuclear import and stability of GATA-3 as a central rheostat for lymphocyte identity
The project addresses the “Re-Thinking Health” concept by focusing on the molecular mechanisms that actively maintain a healthy, stable cellular identity. The dynamic balance of GATA-3’s nuclear import and degradation is a prime example of a health-preserving signalling network (Saikali et al., Cell Rep. 2026). By understanding how this rheostat is finely tuned, we can define the molecular signature of a healthy immune state, providing a critical foundation for recognizing the earliest deviations toward maladaptive, inflammatory conditions and identifying novel targets for prevention.
Project description
The transcription factor GATA-3 is a pillar of lymphocyte biology, orchestrating the development and function of T helper (Th) cells and innate lymphoid cells (ILCs). While the control of GATA-3 expression is well-documented, its activity is also governed by more subtle, dynamic mechanisms. Our recent work has uncovered a critical, previously underappreciated layer of regulation: the precise control of GATA-3’s subcellular localization (Saikali et al., Cell Rep. 2026).
Using quantitative single-cell imaging, we revealed that GATA-3 compartmentalization is tightly regulated in various lymphocyte subsets. In progenitor cells and functional type 2 lymphocytes (Th2, ILC2), GATA-3 is actively concentrated in the nucleus. In stark contrast, type 1 lymphocytes (Th1, ILC1) sequester the majority of GATA-3 protein in the cytoplasm. This spatial segregation is not a static feature but a dynamic rheostat; reprogramming a cell’s identity from type 1 to type 2 forces GATA-3 into the nucleus, and vice-versa, with direct consequences on cellular function. We identified importin-β as the general transporter for GATA-3’s nuclear entry and discovered that nuclear GATA-3 has a remarkably short half-life, necessitating continuous import to maintain Th2 cell identity (Saikali et al., Cell Rep. 2026).
This raises a fundamental question: what is the molecular machinery that enforces this differential localization? The simple presence of importin-β is not sufficient to explain why GATA-3 import is highly efficient in Th2 cells but restricted in Th1 cells. This knowledge gap points to the existence of cell-type-specific signals that fine-tune GATA-3’s transport and stability. Therefore, we posit a central hypothesis: the subcellular localization of GATA-3 is governed by specific post-translational modifications (PTMs) that modulate its affinity for the nuclear import machinery, and subsequently control its degradation, thereby defining the functional identity and stability of lymphocyte lineages. This project aims to dissect these precise molecular mechanisms.
Aim 1: Elucidate the molecular signals governing the differential nuclear import of GATA-3 in type 1 and type 2 lymphocytes. Our data suggest that the affinity of GATA-3 for the importin-β transport machinery is regulated differently in Th1 and Th2 cells. This aim will identify the specific molecular modifications and interacting partners that control the preferential nuclear or cytoplasmic accumulation of GATA-3.
Aim 2: Define the mechanisms controlling the compartmentalized stability of GATA-3. Our finding that nuclear GATA-3 is rapidly degraded while cytoplasmic GATA-3 is more stable is a key component of the regulatory rheostat, as it makes Th2 cells highly dependent on continuous nuclear import. This aim will identify the molecular machinery responsible for the differential stability of GATA-3 in the nucleus versus the cytoplasm.
References
- Saikali P, Dzamukova M, Stehle C, Nguyen TV, Brunner TM, Baumann C, Hegazy AN, Kaufmann SHE, Romagnani C, Löhning M. GATA-3 localization shapes lymphocyte function. Cell Rep. 2026; 45(2): 116925.
Scientific interest within the context of the graduate college
PROJECT 2. Elucidating the structural basis and functional relevance of GATA-3 foci in lymphocytes
Our project is aligned with the “Re-Thinking Health” mission by investigating a fundamental molecular mechanism that defines a healthy, functional state in key immune cells. Rather than focusing on a disease state, the project seeks to understand how the specific nuclear architecture of GATA-3 contributes to the maintenance of immunological balance (Saikali et al., Cell Rep. 2026). This work aims to characterize a molecular hallmark of health, providing a new perspective on how cellular wellness is actively maintained.
Project desciption
GATA-3 is a master-regulator transcription factor essential for the development, functional specification, and homeostasis of numerous cell types. Its role is particularly prominent in the immune system, where it governs the fate and function of T helper 2 (Th2) cells and Group 2 Innate Lymphoid Cells (ILC2s), cell types critically involved in allergic inflammation, anti-helminth immunity, and tissue repair. GATA-3 orchestrates complex gene expression programs by directly binding to DNA and recruiting various co-factors, including other signal transducers, transcription factors, and epigenetic modifiers. Given its central role, the precise regulation of GATA-3 activity is paramount for cellular identity and function, and its dysregulation is a hallmark of diseases such as asthma, allergy, and certain cancers.
While much research has focused on the regulation of GATA-3 expression levels, a fundamental aspect of its biology remains unexplored: its spatial organization within the cell nucleus. Our preliminary work has uncovered a novel phenomenon in select lymphocyte populations. We observed that GATA-3 exhibits two distinct modes of nuclear aggregation: a “diffuse” pattern, where it is distributed throughout the nucleoplasm, and a “punctate” pattern, characterized by the formation of distinct GATA-3 foci in addition to the diffuse staining. The appearance of this punctate pattern is cell-type specific, suggesting it is a tightly regulated process. This observation leads to our hypothesis: the formation of a GATA-3 nuclear focus represents a higher-order regulatory hub that drives unique transcriptional programs, thereby imparting specific functional characteristics to the cell.
We propose that this unique aggregation pattern is not a random event but a key mechanism for modulating GATA-3 activity. The formation of a focus could concentrate GATA-3 and its co-factors, increase its residency time on specific target genes, or sequester it away from others, thus representing a novel layer of gene regulation. This project aims to dissect the molecular basis of this phenomenon and its direct impact on lymphocyte function.
Aim 1: Define the molecular basis underlying the distinct GATA-3 aggregation patterns. To understand why GATA-3 forms these foci, we must first characterize their composition and the conditions that lead to their formation. This aim will investigate the molecular identity of the GATA-3 foci and the cellular machinery that governs their assembly and disassembly.
Aim 2: Manipulate the aggregation pattern and define the functional consequences on cells. The ultimate test of our hypothesis is to control the formation of GATA-3 foci and observe the resulting cellular and molecular changes. This aim will establish a cause-and-effect relationship between GATA-3 aggregation and lymphocyte function.
References
- Saikali P, Dzamukova M, Stehle C, Nguyen TV, Brunner TM, Baumann C, Hegazy AN, Kaufmann SHE, Romagnani C, Löhning M. GATA-3 localization shapes lymphocyte function. Cell Rep. 2026; 45(2): 116925.
Immunometabolic characterization of Crohn’s disease phenotypes – Does location matter?
Principal Investigator

Dr. Lea-Maxie Haag
Dr. Malte Lehmann
Scientific interest within the context of the graduate college
Based at the Department of Gastroenterology, Infectious Diseases and Rheumatology at Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin, our main clinical and research focus are inflammatory bowel diseases (IBD). Crohn’s disease (CD), one of the primary forms of IBD, exhibits various clinical phenotypes, including inflammatory (B1), stricturing (B2), and fistulizing (B3) disease phenotypes. We aim at understanding the biological differences between these phenotypes at the level of the mucosal microenvironment. With our large endoscopy unit and outpatient clinic, as well as gastroenterology ward, we have access to biological samples like intestinal biopsies along with the corresponding clinical data.
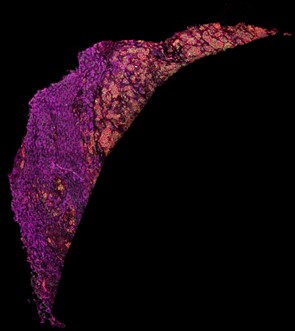
Figure 1. Imaging mass cytometry staining of a liver sample stained with antibodies directed against fatty acid synthase (magenta), voltage-dependent anion-channel 1 (green) and CD44 (red).
Project description
What is the underlying mechanism and biology leading to the different disease locations in Crohn’s disease? This project aims to characterize the immunometabolic landscape of Crohn’s disease depending on disease location. Using an already established 35-marker imaging mass cytometry (IMC) panel, we will analyze mucosal tissue from patients with ileal, colonic, and ileocolonic Crohn’s disease and compare these samples with ulcerative colitis and healthy controls. This approach enables simultaneous analysis of immune, epithelial, and stromal cells at single-cell resolution within their spatial tissue context. In addition to identifying cell populations, it allows investigation of metabolic states, cell-cell interactions, and cellular neighborhoods within the intestinal mucosa.
The project builds on extensive preliminary work from our group, including established IMC workflows, published expertise in spatial single-cell profiling, and an already collected and well-characterized cohort. The required FFPE samples and corresponding clinical data are already available, and all relevant analytical methods are established, making this a highly feasible and well-supported research project.
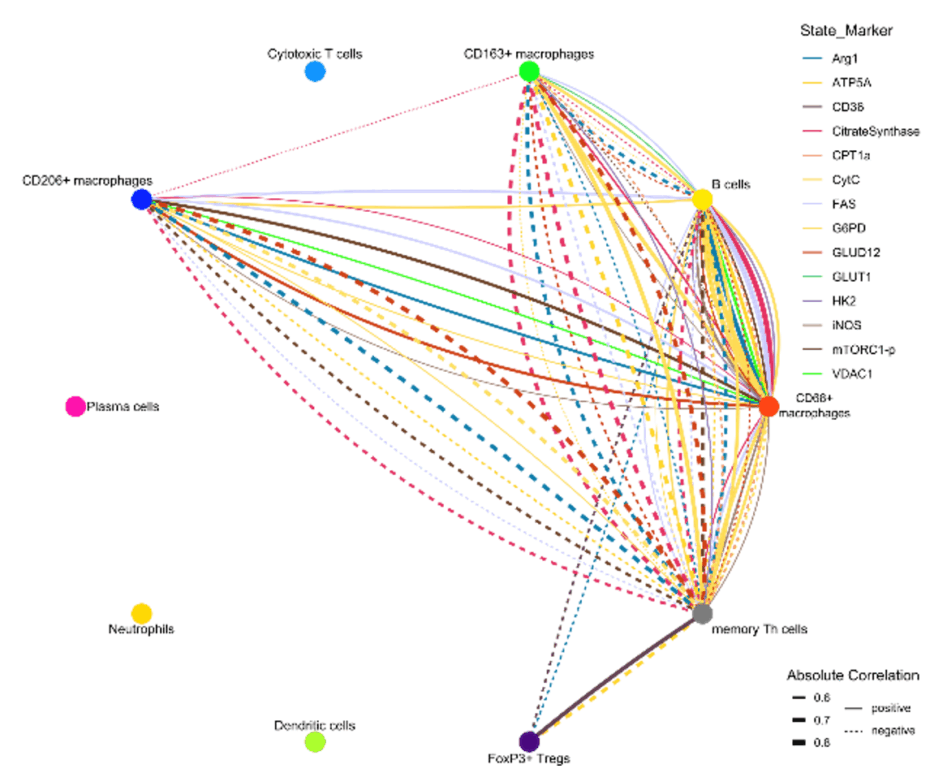
Figure 2. Iconography of correlations of state marker expression to the distance between immune cell clusters. Only correlations with r > 0.5 or < 0.5 and p < 0.05 are shown. The color of the lines indicates the respective state marker, the width of the line the absolute value of correlation. Straight lines indicate positive correlations, doted lines negative correlations.
WP1: Cohort familiarization and sample annotation. Introduction to the clinical cohort and classification of samples from ileal, colonic, and ileocolonic Crohn’s disease, ulcerative colitis, and healthy controls.
WP2: Imaging mass cytometry data generation and analysis. Participation in IMC-based analysis of mucosal biopsies using an established 35-marker panel.
WP3: Spatial and immunometabolic profiling. Analysis of immune cell composition, epithelial and stromal cell states, metabolic marker expression, cell-cell interactions and tissue neighborhoods, differences between disease locations and disease entities.
What the student will do: i) work on a translational IBD research project addressing an important clinical question; ii) analyze high-dimensional spatial single-cell data from human intestinal tissue; iii) compare immunometabolic tissue signatures across Crohn’s disease locations; iv) contribute to interpretation of findings in the context of disease mechanisms and therapy response; v) participate in scientific discussion, presentation of results, and manuscript development.
What the student will learn: i) principles of translational inflammatory bowel disease research in Gastroenterology; ii) application and interpretation of imaging mass cytometry data, iii) spatial single-cell and immunometabolic tissue analysis; iv) R-based analysis workflows for multiplex imaging data; v) scientific writing, presentation, and work in an interdisciplinary clinician-scientist environment.
Supervision and environment. The student will be embedded in an experienced and collaborative research team with close supervision by clinicians and clinician-scientists working at the interface of patient care and experimental research. The group has extensive expertise in inflammatory bowel disease, tissue immunology, and imaging mass cytometry. Weekly meetings, scientific exchange, and structured mentoring will support the student throughout the project.
Expected outcome: i) completion of a MD thesis; ii) strong potential for co-authorship or first authorship, depending on progress and project scope; iii) acquisition of valuable skills for a future clinical-academic career in gastroenterology or immunology.
Target group. This project is ideally suited for a highly motivated medical student seeking a one-year full-time research experience and interested in translational medicine, gastroenterology, immunology, and advanced tissue imaging.
References
- Gajendran M, Loganathan P, Catinella AP, Hashash JG. A comprehensive review and update on Crohn’s disease. Dis Mon. 2018; 64(2): 20-57.
- Atreya R, Siegmund B. Location is important: differentiation between ileal and colonic Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2021; 18(8): 544-558.
- Lehmann M, Weixler B, Elezkurtaj S, Loddenkemper C; TRR241 IBDome Consortium; Kühl AA, Siegmund B. Spatial single cell profiling using imaging mass cytometry: inflammatory versus penetrating Crohn’s disease. J Crohns Colitis. 2024; 18(8): 1305-1318.
- Lehmann M, Allers K, Heldt C, Meinhardt J, Schmidt F, Rodriguez-Sillke Y, Kunkel D, Schumann M, Böttcher C, Stahl-Hennig C, Elezkurtaj S, Bojarski C, Radbruch H, Corman VM, Schneider T, Loddenkemper C, Moos V, Weidinger C, Kühl AA, Siegmund B. Human small intestinal infection by SARS-CoV-2 is characterized by a mucosal infiltration with activated CD8+ T cells. Mucosal Immunol. 2021; 14(6): 1381-1392.
- Stankey CT*, Bourges C*, Haag LM*, Turner-Stokes T, Piedade AP, Palmer-Jones C, Papa I, Silva Dos Santos M, Zhang Q, Cameron AJ, Legrini A, Zhang T, Wood CS, New FN, Randzavola LO, Speidel L, Brown AC, Hall A, Saffioti F, Parkes EC, Edwards W, Direskeneli H, Grayson PC, Jiang L, Merkel PA, Saruhan-Direskeneli G, Sawalha AH, Tombetti E, Quaglia A, Thorburn D, Knight JC, Rochford AP, Murray CD, Divakar P, Green M, Nye E, MacRae JI, Jamieson NB, Skoglund P, Cader MZ, Wallace C, Thomas DC, Lee JC. A disease-associated gene desert directs macrophage inflammation through ETS2. Nature. 2024 Jun; 630(8016): 447-456. *equal contribution
- Avery EG*, Haag LM*, McParland V, Kedziora SM, Zigra GJ, Valdes DS, Kirchner M, Popp O, Geisberger S, Nonn O, Karlsen TV, N’Diaye G, Yarritu A, Bartolomaeus H, Bartolomaeus TUP, Tagiyeva NA, Wimmer MI, Haase N, Zhang YD, Wilhelm A, Grütz G, Tenstad O, Wilck N, Forslund SK, Klopfleisch R, Kühl AA, Atreya R, Kempa S, Mertins P, Siegmund B, Wiig H, Müller DN. Intestinal interstitial fluid isolation provides novel insight into the human host-microbiome interface. Cardiovasc Res. 2025; 121(5): 803-816. *equal contribution
Epithelial acetyl-CoA metabolism as a regulator of intestinal barrier function in IBD
Prinicipal Investigator

Prof. Dr. Britta Siegmund
Dr. Lea-Maxie Haag
Dr. Malte Lehmann
Scientific interest within the context of the graduate college
Inflammatory bowel disease (IBD) is a chronic, multifactorial disorder characterized by immune dysregulation, microbial imbalance, and impaired epithelial barrier function. Intestinal epithelial cells (IECs) play a central role in maintaining gut homeostasis by integrating signals from the microbiota and the immune system. Emerging evidence from immunometabolism highlights that metabolic reprogramming is a key regulator of inflammatory processes. However, IEC-intrinsic metabolic alterations in IBD remain poorly understood.
Within our preliminary work, integrative analyses of human IBD datasets and tissue samples revealed profound metabolic dysregulation in IECs, with the acetyl-CoA biosynthesis pathway emerging as a critical metabolic hub. In particular, reduced expression of key enzymes such as ACLY and ACSS2 suggests that impaired epithelial metabolism may compromise barrier integrity and promote inflammation. Together, these findings support the concept that epithelial metabolic rewiring is a central driver of IBD pathogenesis and a promising target for therapeutic intervention. Our research group at the Department of Gastroenterology, Infectious Diseases and Rheumatology at Charité – Universitätsmedizin Berlin (Campus Benjamin Franklin) and the Max Delbrück Center (MDC) conducts translational research in IBD, leveraging access to a large, deeply phenotyped patient cohort and comprehensive biobanking infrastructure. The project is embedded within the Collaborative Research Centre TRR241, which integrates clinical, experimental, and computational approaches within the IBDome framework to systematically dissect mechanisms of intestinal inflammation. Within this network, we contribute the human translational component, including access to patient samples and multi-omic analyses, while a partner laboratory in Erlangen performs complementary mechanistic studies in murine models. This integrated, cross-site approach enables the direct linking of human disease phenotypes with experimentally defined mechanisms.
Project description
This project aims to define how epithelial metabolic rewiring contributes to intestinal inflammation in human inflammatory bowel disease (IBD), with a particular focus on the acetyl-CoA biosynthesis pathway. Building on our preliminary findings identifying ACLY and ACSS2 as key metabolic regulators in IECs, we will investigate how altered epithelial metabolism affects barrier integrity and mucosal homeostasis in patients. Using biopsies from well-characterized IBD cohorts, we will perform metabolomic profiling of primary IECs and apply stable isotope tracing to reconstruct metabolic pathway activity. Patient-derived intestinal organoids will be used to distinguish epithelial-intrinsic changes from microenvironment-driven effects and to assess responses to inflammatory and microbial stimuli. In parallel, spatial proteomics and interstitial fluid analysis will provide insight into the local signaling environment shaping epithelial metabolism. By integrating these approaches, the project aims to identify key metabolic pathways underlying epithelial dysfunction in IBD and reveal potential targets for therapeutic intervention.
Embedded within the Collaborative Research Centre TRR241, access to well-characterized patient cohorts, high-quality clinical data, and biological samples is assured. All experimental protocols are established, and data acquisition is ongoing. The existing research network, dedicated study teams, and continuous patient recruitment provide a strong infrastructure and ensure close supervision and reliable project execution.
WP1: Characterization of acetyl-CoA pathway components in human IBD. We will analyze ACLY and ACSS2 expression, localization, and phosphorylation in intestinal biopsies from IBD patients across disease states. Using immunostaining, Western blotting, and integration of IBDome and scRNA-seq datasets, we will correlate epithelial acetyl-CoA pathway activity with disease type, inflammation grade, and therapeutic response.
WP2: Functional analysis of epithelial acetyl-CoA metabolism. Patient-derived intestinal organoids will be used to model epithelial-intrinsic metabolic alterations. Pharmacological inhibition of ACLY and ACSS2 will mimic loss-of-function, and metabolic consequences will be assessed by selected metabolic readouts. In parallel, targeted metabolomic profiling will characterize key acetyl-CoA–related pathways in IBD.
WP3: Spatial characterization of epithelial metabolic rewiring in IBD. We will perform spatial proteomics on FFPE samples from the IBDome cohort to map epithelial protein expression in situ, focusing on UC pancolitis stratified by inflammation status. Laser microdissection followed by mass spectrometry will enable analysis of protein abundance and key post-translational modifications.To complement this, isotope-resolved metabolomics will be applied to selected biopsies to assess acetyl-CoA–related metabolic activity. In addition, interstitial fluid analysis will characterize the local signaling environment, including cytokines and metabolites.
Together, these approaches will provide a spatially resolved view of epithelial metabolism and its regulation within the intestinal microenvironment.
What the student will do: i) work on a translational research project investigating epithelial metabolism in human IBD; ii) analyze human intestinal biopsies, primary epithelial cells, and patient-derived organoids; iii) perform and interpret metabolomic and spatial proteomic analyses; iv) contribute to the characterization of metabolic alterations across disease states; v) participate in data integration, scientific discussion, and manuscript preparation.
What the student will learn: i) principles of translational research in IBD and mucosal immunology; ii) hands-on experience with metabolomics, isotope tracing, and spatial proteomics; iii) analysis and interpretation of multi-omic datasets; iv) scientific writing, presentation skills, and critical data interpretation; v) working in an interdisciplinary clinician-scientist environment.
Supervision and environment. The student will be embedded in an experienced and collaborative research team at Charité and MDC, with close supervision by clinician scientists. The project is part of the CRC241 research network, providing access to a multidisciplinary environment integrating clinical, experimental, and computational expertise. Regular meetings, seminars, and structured mentoring will support the student throughout the project.
Expected outcome. i) completion of a MD thesis; ii) strong potential for co-authorship or first authorship, depending on progress; iii) acquisition of key skills for a future clinical-academic career.
Target group. This project is aimed at highly motivated and academically outstanding medical students seeking a dedicated one-year full-time research experience. Candidates should have a strong interest in translational medicine, gastroenterology, and immunometabolism, as well as a willingness to engage with advanced experimental and analytical approaches. Prior research experience is advantageous but not required. A high level of motivation, reliability, and curiosity is essential.
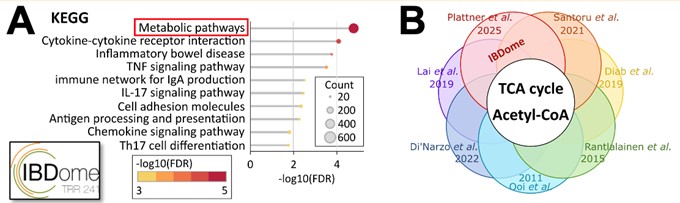
Figure 1. (A) KEGG pathway analysis of differentially expressed genes between IBD patients and controls (IBDome cohort, TRR241). (B) Schematic of overlapping pathways reported in the literature.
References
- Stankey CT*, Bourges C*, Haag LM*, Turner-Stokes T, Piedade AP, Palmer-Jones C, Papa I, Silva Dos Santos M, Zhang Q, Cameron AJ, Legrini A, Zhang T, Wood CS, New FN, Randzavola LO, Speidel L, Brown AC, Hall A, Saffioti F, Parkes EC, Edwards W, Direskeneli H, Grayson PC, Jiang L, Merkel PA, Saruhan-Direskeneli G, Sawalha AH, Tombetti E, Quaglia A, Thorburn D, Knight JC, Rochford AP, Murray CD, Divakar P, Green M, Nye E, MacRae JI, Jamieson NB, Skoglund P, Cader MZ, Wallace C, Thomas DC, Lee JC. A disease-associated gene desert directs macrophage inflammation through ETS2. Nature. 2024; 630(8016): 447-456. *equal contribution
- Avery EG*, Haag LM*, McParland V, Kedziora SM, Zigra GJ, Valdes DS, Kirchner M, Popp O, Geisberger S, Nonn O, Karlsen TV, N’Diaye G, Yarritu A, Bartolomaeus H, Bartolomaeus TUP, Tagiyeva NA, Wimmer MI, Haase N, Zhang YD, Wilhelm A, Grütz G, Tenstad O, Wilck N, Forslund SK, Klopfleisch R, Kühl AA, Atreya R, Kempa S, Mertins P, Siegmund B, Wiig H, Müller DN. Intestinal interstitial fluid isolation provides novel insight into the human host-microbiome interface. Cardiovasc Res. 2025; 121(5): 803-816. *equal contribution
- Santoru ML, Piras C, Murgia F, Leoni VP, Spada M, Murgia A, Liggi S, Lai MA, Usai P, Caboni P, Manzin A, Atzori L. Metabolic alteration in plasma and biopsies from patients with IBD. Inflamm Bowel Dis. 2021; 27(8): 1335-1345.
- Diab J, Hansen T, Goll R, Stenlund H, Jensen E, Moritz T, Florholmen J, Forsdahl G. Mucosal metabolomic profiling and pathway analysis reveal the metabolic signature of ulcerative colitis. Metabolites. 2019; 9(12): 291.
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, Sud M, Andrews E, Velonias G, Haber AL, Jagadeesh K, Vickovic S, Yao J, Stevens C, Dionne D, Nguyen LT, Villani AC, Hofree M, Creasey EA, Huang H, Rozenblatt-Rosen O, Garber JJ, Khalili H, Desch AN, Daly MJ, Ananthakrishnan AN, Shalek AK, Xavier RJ, Regev A. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell. 2019; 178(3): 714-730.e22.
Immunological health: the role of EPHB3 in enabling and preserving human T-cell immunity
Prinicipal Investigator

Prof. Dr. Horst von Bernuth
Scientific interest within the context of the graduate college
Health is not merely the absence of disease, but a dynamic, active molecular process where the development and preservation of a competent immune system is crucial. A core characteristic of immunological health is thymopoiesis. Impaired development or decay of thymopoietic capacity shorten the health span through increased susceptibility to infectious diseases, cancer, autoimmunity, and inflammatory disorders.1,2 To extend the human health span, we need to define the homeostatic pathways that actively enable and preserve immunological health. Still, crucial signaling pathways governing T-cell development in the thymus are insufficiently characterized on the molecular level in humans. Such insights are important for the early detection of deviations from health for diagnostic purposes and for the development of intervention strategies to re-establish healthy thymopoiesis.1,3
Patients with rare mutations in key pathways for the development and function of both the thymus stromal compartment and the lymphoid compartment offer a unique model to address this knowledge gap. We aim to identify the health-enabling signaling networks required to build and preserve thymus-dependent T-cell homeostasis as a foundation for future diagnostics and interceptive therapies to establishing and maintaining immunological health.
Project description
This project aims at defining the functional contribution of EPHB3 to immunological health, using a newly identified mutation in combined immunodeficiency (CID) patients. EPHB3 is a Receptor Tyrosine Kinase (RTK) capable of bidirectional signaling.4 While murine knock-out models (Ephb3−/−) demonstrate T-cell lymphopenia and premature immunosenescence as a consequence of impaired thymopoiesis, its role in human remains unknown.5 We have identified two patients from a single family (mother and son) presenting with a variant in the ligand-binding domain (LBD) of the EPHB3 gene. One patient exhibits autoimmunity with early-onset juvenile idiopathic arthritis and there is infection susceptibility in the family history with even an early fatal pneumonia, mirroring the murine phenotype and indicating dysfunctional thymopoiesis. Accordingly, the immunological phenotype displays a reduction in T-, B- and NK cells and thus, is classified as CID. This represents the first-in-human description of a disease-causing variant in EPHB3, offering the unique opportunity to study the role of EPHB3 in human immunological health. According to available data, EPHB3 is expressed within thymic epithelial cells (TECs), suggesting a critical role in the thymic stroma.6 However, our own preliminary data using artificial thymic organoids (ATOs) in vitro display an additional cell-intrinsic defect in developing T-cell precursors, pointing towards a previously not described, T-cell intrinsic function of EPHB3. We hypothesize that the variant impairs ligand binding and signal transduction, thereby disrupting the critical ligand-receptor crosstalk required for development of both thymic stroma cells and T-cells.
Therefore, the MD candidate will assess the functional role of EPHB3 for both thymic stroma cells as well as T-cell precursors and characterize the impact of the newly identified mutation (Figure 1). Specifically, in work package 1 (WP1), the candidate will conduct a functional validation of the mutation (by RT-PCR, Western blotting, and phosphorylation assays) using primary patient samples (PBMCs) as well as edited cell lines generated by the candidate (Ephb3−/− HEK 293T cell line with overexpression plasmids coding for wild type vs. patient variant). In WP2, the candidate will assess the functional contribution of EPHB3 for T lymphocytes maturation and functionality by transcriptomic (RNAseq) and phenotypic (FACS) profiling of patient-derived and control T-cell subsets ex vivo as well as of patient and control HSC-derived T-cell precursors generated in ATOs in vitro.7 To assess the role of EPHB3 for thymic stroma cells (WP3), the candidate will leverage an in vitro iPSC to thymus epithelial progenitor cell differentiation protocol. The required (patient-derived) iPSC lines, will be generated by the BIH Core Unit pluripotent Stem Cells and Organoids (CUSCO).8,9 To assess a possible role of EPHB3 for premature immunosenescence of T-cells (WP4), the candidate will conduct profiling (epigenetic, genomic, and phenotypic) for markers of cellular senescence in primary T cells from the patients and healthy controls.10,11 The candidate will also assess autoantibodies in the sera of patients and controls, as an indicators of premature thymic involution.12,13,14 Embedded in the Else Kröner-Promotionskolleg Re-Thinking Health, this project enables a first-in-human description of the function of EPHB3 for immunological health with future translational capacity.
Abbreviations: HSCs, hematopoietic stem cells; LBD, ephrin ligand-binding domain; WP, work package
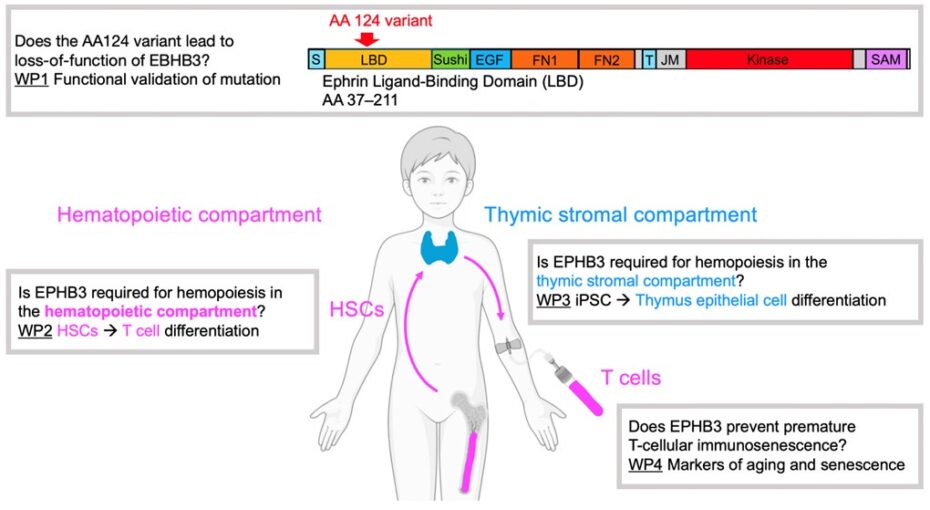
Figure 1. Visual project summary.
References
- Dinges SS, Amini K, Notarangelo LD, Delmonte OM. Primary and secondary defects of the thymus. Immunol Rev. 2024; 322(1): 178-211.
- Liu Z, Liang Q, Ren Y, Guo C, Ge X, Wang L, Cheng Q, Luo P, Zhang Y, Han X. Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther. 2023; 8(1): 200.
- Kreins AY, Maio S, Dhalla F. Inborn errors of thymic stromal cell development and function. Semin Immunopathol. 2021; 43(1): 85-100.
- Pasquale EB. Eph receptor signaling complexes in the plasma membrane. Trends Biochem Sci. 2024; 49(12): 1079-1096.
- García-Ceca J, Montero-Herradón S, Zapata AG. Thymus aging in mice deficient in either EphB2 or EphB3, two master regulators of thymic epithelium development. Dev Dyn. 2020; 249(10): 1243-1258.
- Speir ML, Bhaduri A, Markov NS, Moreno P, Nowakowski TJ, Papatheodorou I, Pollen AA, Raney BJ, Seninge L, Kent WJ, Haeussler M. UCSC cell browser: visualize your single-cell data. Bioinformatics. 2021; 37(23): 4578-4580.
- Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, Sacchetti N, Kawai T, Garabedian EK, Draper D, Bergerson JRE, DeRavin SS, Freeman AF, Güngör T, Hartog N, Holland SM, Kohn DB, Malech HL, Markert ML, Weinacht KG, Villa A, Seet CS, Montel-Hagen A, Crooks GM, Notarangelo LD. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv. 2020; 4(12): 2611-2616.
- Provin N, d’Arco M, Le Bozec A, Kervagoret E, Bruneau A, Brusselle L, Fourgeux C, Poschmann J, Hulin P, Maminirina P, Baron O, Saulquin X, Guillonneau C, David L, Giraud M. Combinatory differentiation of human induced pluripotent stem cells generates functional thymic epithelium driving dendritic cell and CD4/CD8 T-cell development. Nat Commun. 2026; 17(1): 1969.
- Yamazaki Y, Urrutia R, Franco LM, Giliani S, Zhang K, Alazami AM, Dobbs AK, Masneri S, Joshi A, Otaizo-Carrasquero F, Myers TG, Ganesan S, Bondioni MP, Ho ML, Marks C, Alajlan H, Mohammed RW, Zou F, Valencia CA, Filipovich AH, Facchetti F, Boisson B, Azzari C, Al-Saud BK, Al-Mousa H, Casanova JL, Abraham RS, Notarangelo LD. PAX1 is essential for development and function of the human thymus. Sci Immunol. 2020; 5(44): eaax1036.
- Xu W, Larbi A. Markers of T-cell senescence in humans. Int J Mol Sci. 2017; 18(8): 1742.
- Thewissen M, Somers V, Venken K, Linsen L, van Paassen P, Geusens P, Damoiseaux J, Stinissen P. Analyses of immunosenescent markers in patients with autoimmune disease. Clin Immunol. 2007; 123(2): 209-218.
- Staudacher O, Meyer T, Akbil B, Mayer M, Schmoll C, Kölsch U, Unterwalder N, Slagman A, Meisel C, Goffinet C, Möckel M, von Bernuth H. Autoantibodies against type I interferons correlate with low CD169/SIGLEC1 and severe non-viral infections in ER patients. Clin Exp Immunol. 2026; 220(1): uxaf074.
- Meisel C, Akbil B, Meyer T, Lankes E, Corman VM, Staudacher O, Unterwalder N, Kölsch U, Drosten C, Mall MA, Kallinich T, Schnabel D, Goffinet C, von Bernuth H. Mild COVID-19 despite autoantibodies against type I IFNs in autoimmune polyendocrine syndrome type 1. J Clin Invest. 2021; 131(14): e150867.
- Akbil B, Meyer T, Stubbemann P, Thibeault C, Staudacher O, Niemeyer D, Jansen J, Mühlemann B, Doehn J, Tabeling C, Nusshag C, Hirzel C, Sanchez DS, Nieters A, Lother A, Duerschmied D, Schallner N, Lieberum JN, August D, Rieg S, Falcone V, Hengel H, Kölsch U, Unterwalder N, Hübner RH, Jones TC, Suttorp N, Drosten C, Warnatz K, Spinetti T, Schefold JC, Dörner T, Sander LE, Corman VM, Merle U; Pa-COVID study Group; Kurth F, von Bernuth H, Meisel C, Goffinet C. Early and rapid identification of COVID-19 patients with neutralizing type I interferon auto-antibodies. J Clin Immunol. 2022; 42(6): 1111-1129.
Plasticity of ILC3 in intestinal inflammation
Principle Investigator

Dr. Michael Kofoed-Branzk
Scientific interest within the context of the graduate college
We study the development and function of the innate immune system, with a particular emphasis on innate lymphoid cells (ILC). Our current goal is to achieve a molecular-level understanding of how the innate immune system integrates environmental cues (such as those derived from nutrients, microbiota, and circadian rhythm) to influence tissue physiology. Recent findings have uncovered increasingly complex interactions between components of the innate immune system and fundamental developmental and biological processes, pointing to previously unrecognized pathways through which immune mechanisms may be harnessed to enhance health and longevity. These insights suggest expanded roles for the immune system in regulating tissue homeostasis, morphogenesis, metabolism, regeneration, and growth. Our work is inherently interdisciplinary, bridging fields such as immunology, microbiology, epigenetics, developmental and stem cell biology, nutrition science, tumor biology, and regenerative medicine.
Project description
Group 3 Innate lymphoid cells (ILC3) are tissue-resident innate lymphocytes that are involved in immunity to infections but are also deeply integrated in the regulation of tissue function. ILC3 are crucial regulators of intestinal barrier homeostasis in both health and disease. In contrast to adaptive lymphocytes, they are constantly active and serve both as direct responders, i.e. by sensing mediators and metabolites and secreting effector cytokines, but also participate in discrete regulatory circuits with both immune and non-hematopoietic cells. However, ILC3 are highly heterogenous, and different subpopulations have markedly different phenotypes with differential expression of immune regulatory receptors and effector cytokines. Recent findings from our laboratory indicate that this heterogeneity arises due to post-developmental plasticity of ILC3 in response to changes in the tissue (micro)environment. Specifically, we have demonstrated that ILC3 are epigenetically poised for alternate cell fate decisions, resulting in subsequent changes in effector functions (manuscript in preparation). Importantly, our data suggests that this epigenetically encoded plastic potential may constitute an intrinsic switch that enable ILC3 to adapt during intestinal inflammation, as an alternative mode of defense to reestablishment of barrier integrity and homeostasis.
In a TNF-driven model of inflammatory bowel disease (IBD), we observe a transformation of the ILC3 compartment indicating that such epigenetic reprogramming is occurring. The drivers of the change of the intestinal ILC3 compartment, and the immediate and long-term consequences remain to be elucidated. We hypothesize that intestinal inflammation activates these epigenetically encoded programs in ILC3 and that the propensity for such reprogramming is further regulated by both intrinsic and extrinsic factors (e.g. cytokines, vitamins and aryl hydrocarbons).
In this proposal, we aim to investigate the epigenetic foundation of ILC3 lineage stability in the context of intestinal inflammation, whether this is causally linked to disease progression and explore if targeted intervention of ILC3 plasticity may affect disease progression and outcome.
Aim 1: Characterize the lineage stability and plasticity of ILC3 in murine models of intestinal bowel disease. We will use established protocols for epigenetic profiling of ILC3 combined with transcriptomic analysis and state of the art lineage tracing techniques to identify changes in ILC3 during intestinal inflammation.
Aim 2: Investigate the cause-and-effect relationships of ILC3 plasticity in intestinal inflammation. Using both novel and established genetic targeting of ILC3 as well as dietary interventions, we will investigate the causal relationships of ILC3 plasticity in intestinal inflammation.
References
- Biniaris-Georgallis SI, Aschman T, Stergioula K, Schreiber F, Jafari V, Taranko A, Karmalkar T, Kasapi A, Lenac Rovis T, Jelencic V, Bejarano DA, Fabry L, Papacharalampous M, Mattiola I, Molgora M, Hou J, Hublitz KW, Heinrich F, Guerra GM, Durek P, Patone G, Lindberg EL, Maatz H, Hölsken O, Krönke G, Mortha A, Voll RE, Clarke AJ, Hauser AE, Colonna M, Thurley K, Schlitzer A, Schneider C, Stamatiades EG, Mashreghi MF, Jonjic S, Hübner N, Diefenbach A*, Kanda M*, Triantafyllopoulou A*. Amplification of autoimmune organ damage by NKp46-activated ILC1s. Nature. 2024; 634(8035): 952-960. *equally contributing senior authors
- Guendel F, Kofoed-Branzk M, Gronke K, Tizian C, Witkowski M, Cheng HW, Heinz GA, Heinrich F, Durek P, Norris PS, Ware CF, Ruedl C, Herold S, Pfeffer K, Hehlgans T, Waisman A, Becher B, Giannou AD, Brachs S, Ebert K, Tanriver Y, Ludewig B, Mashreghi MF, Kruglov AA, Diefenbach A. Group 3 innate lymphoid cells program a distinct subset of IL-22BP-producing dendritic cells demarcating solitary intestinal lymphoid tissues. Immunity. 2020; 53(5): 1015-1032.e8.
- Schaupp L, Muth S, Rogell L, Kofoed-Branzk M, Melchior F, Lienenklaus S, Ganal-Vonarburg SC, Klein M, Guendel F, Hain T, Schütze K, Grundmann U, Schmitt V, Dorsch M, Spanier J, Larsen PK, Schwanz T, Jäckel S, Reinhardt C, Bopp T, Danckwardt S, Mahnke K, Heinz GA, Mashreghi MF, Durek P, Kalinke U, Kretz O, Huber TB, Weiss S, Wilhelm C, Macpherson AJ, Schild H, Diefenbach A1,*, Probst HC*. Microbiota-induced type I interferons instruct a poised basal state of dendritic cells. Cell. 2020; 181(5): 1080-1096.e19. 1lead senior author, *equally contributing senior authors
- Gronke K, Hernández PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, Witkowski M, Tizian C, Amann L, Schumacher F, Glatt H, Triantafyllopoulou A, Diefenbach A. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. 2019; 566(7743): 249-253.
Characterization of long-term immobility induced thromboprotective mechanisms in immune cells
Principle Investigator

Dr. Vincent Ehreiser
Scientific interest within the context of the graduate college
Disease prevention is taking a central role in tackling the burden of cardiovascular disease in our aging society. Thrombotic cardiovascular diseases such as venous thromboembolism, stroke and myocardial infarction are main drivers of morbidity and mortality worldwide. A central pathomechanism in these thrombotic cardiovascular diseases is termed thromboinflammation and arises from a deleterious dysregulation of an evolutionary conserved host defence mechanism involving platelets, the innate immune and coagulation system. There is an unmet clinical need for a better mechanistical understanding of thromboinflammation, in order to novel regulatory mechanisms that may pave the way for potential therapeutics, biomarkers and preventive measures. By pursuing this aim we established a across species approach to investigate the paradox that long-term immobility during hibernation in brown bears and patients with spinal cord injury (SCI) does not increase the risk of thrombosis. Our research acts at the interface between immunology, cardiovascular science and preventive medicine by taking advantage of an interdisciplinary translational research approach.
Project description
Cardiovascular diseases (CVDs), in particular myocardial infarction, stroke and venous thromboembolism (VTE), are the leading cause of morbidity and mortality worldwide, accounting for more than one third of all global deaths. Moreover, thrombotic events frequently and severely complicate a broad spectrum of clinical conditions that extend beyond the classical mechanisms of CVDs. Immobility and bed rest are among the strongest risk factors for VTE with increasing relevance in an aging population. Our previous study has demonstrated a naturally occurring thromboprotective phenotype in hibernating brown bears, bed-resting individuals, and SCI patients.1 This project examines neutrophils as central drivers of a thromboprotective phenotype during prolonged immobilization, employing a cross-species comparative approach that integrates hibernating brown bears and bed-resting human subjects. The overarching aim is to delineate thromboprotective signatures of neutrophils in long-term immobility, thereby uncovering neutrophil-intrinsic pathways that perpetuate thromboinflammatory responses. Building on these characterizations, we will establish the mechanistic foundation for identifying novel therapeutic targets in neutrophil-driven thromboinflammation.
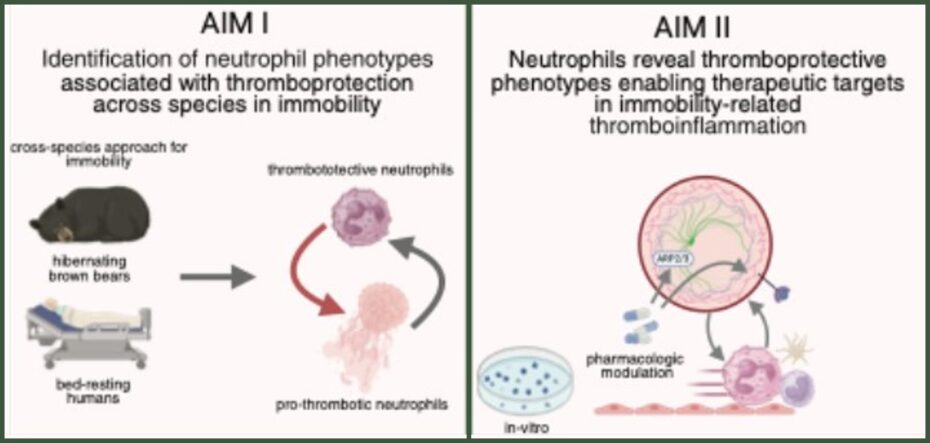
Figure 1. Project scheme illustrative depiction of our cross-species approach to delineate thromboprotective signatures of neutrophils in immobility. Created with biorender.com.
In 2024 we serially captured free ranging brown bears during hibernation and the following activity period as previously described. Isolated neutrophils showed functional alterations, with reduced phagocytic activity reflected by decreased uptake of fluorescently labelled E. coli particles during winter. This indicates an altered neutrophil function between long‑term immobility (Figure 2/A-B), as well as reduced formation of neutrophil extracellular traps (NETs) (Figure 2/C). Extending our initial approach, we assessed neutrophil function in human volunteers undergoing a bed rest protocol at the DLR in Cologne. The functional profile reflects that observed in long‑term immobilized hibernating brown bears, with neutrophils displaying reduced phagocytotic capacities (Figure 1/D) and decreased NET formation in-vitro (Figure 2/E-F).
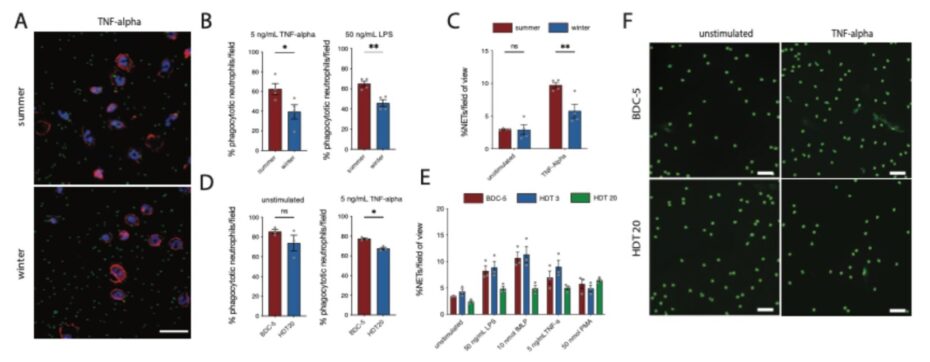
Figure 2. (A) Representative fluorescence microscopy images of neutrophils and labelled E. coli (scale bar: 20 µm). Phagocytosis and NET formation analysis. (B) Percentage of phagocytosing neutrophils in hibernating and active brown bears. (C) Comparative percentage of NET-forming neutrophils in hibernating brown bears. Comparative analysis in bed rest individuals at baseline mobility and after 20 days of bed rest, (D) percentages of phagocytosing and (E) NETing neutrophils. (F) Representative in vitro images of neutrophil NETosis (scale bar: 50 µm).
Quantitative proteomics (Figure 3) delineated differentially regulated proteins in brown bear neutrophils across summer activity and immobility during hibernation in winter. Concordantly, comparative proteomic analysis of neutrophils from our human bed-rest cohort demonstrated immobility-associated downregulation of the ARP2/3 complex as well as associated downstream proteins. The ARP2/3 complex is a key component of the actin cytoskeleton, and its inhibition has previously been shown to impair cellular migration in neutrophils.2-4 Moreover, neutrophils derived from patients with rare inherited defects in actin polymerization, specifically due to deficiencies in the actin-related protein 2/3 complex exhibited a defect for NETosis.5
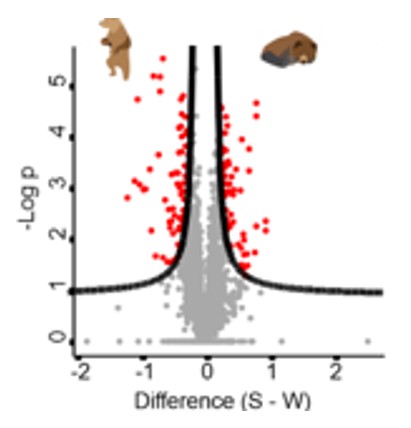
Figure 3. Quantitative proteomic analysis of hibernating and active brown bears. Blue marking for significant proteins with a p-value <0.01.
Aim 1: Identification of neutrophil phenotypes associated with thromboprotection in two models of immobility across species. We hypothesize that neutrophil functions, encompassing phagocytosis, NETosis, actin-dependent spreading, migration, and degranulation are reprogrammed during prolonged immobility in hibernation in brown bears, resulting in a functionally hyporesponsive phenotype in neutrophils that attenuates prothrombotic potential and confers protection against immobility-associated VTE. Within Aim 1, we will delineate these functional alterations through an established collaboration with the Scandinavian Brown Bear Research Project procuring neutrophils from hibernating and active bears in Sweden for on-site in-vitro experiments and validate our previous results. Building on our proteomic analyses, which predominantly identified proteins of the actin‑associated cytoskeleton, we will specifically assess neutrophil migration and cell‑spreading behavior.6 In addition, during the active phase of the brown bears, neutrophils will be treated in vitro with an Arp2/3 complex inhibitor to assess whether their prothrombotic phenotype under conditions of mobility can be shifted toward an antithrombotic phenotype. In analogy to the experiments described in brown bears, we will conduct in vitro experiments as well as further studies within the collaboration of the bed rest study. In analogy to the experiments described in brown bears, we will conduct in vitro experiments as well as further studies within the collaboration of the bed rest study. Cell-free supernatants derived from neutrophil stimulation experiments will be used for proteomic analysis to quantify secreted mediators, including myeloperoxidase (MPO) and neutrophil elastase, thereby correlating degranulation dynamics with antithrombotic phenotype during hibernation. Plasma samples will be subjected to immunoprofiling by ELISA and mass spectrometry-based cytokine arrays to identify pro-inflammatory and thrombogenic mediators, such as interleukins and other DAMPs implicated in neutrophil priming and NETosis in mobility.
Aim 2: Neutrophil crosstalk reveals thromboprotective phenotypes enabling therapeutic targets in immobility-related thromboinflammation. In Aim 2, we will dissect differentially regulated proteins and signalling pathways identified in neutrophils from hibernating brown bears and bed-resting individuals, assessing their capacity to evade short-term immobility-induced proinflammatory phenotypes and promote a thromboprotective state under chronic immobilization. Building on our preliminary data we identified the ARP2/3 complex as a prominently downregulated protein during immobility in neutrophils from hibernating brown bears and human bed-resting individuals. Thus, we will perform functional in-vitro experiments to assess its effect on neutrophil phenotypes, and its influence on the interaction between neutrophils and immune cells (i.e. platelets, monocytes) in the context of thromboinflammation. To this end, we will treat isolated neutrophils with ARP 2/3 inhibitors or vehicle controls and subsequently subject them to phenotypic and functional analysis to assess neutrophil effector functions. Neutrophil functionality will be evaluated using established phagocytosis assays, NETosis quantification, as well as degranulation assays. Additionally, co-culture models (i.e. platelet-neutrophils) will be employed to quantify ARP 2/3 complex inhibition effects on NET release under thromboinflammatory conditions, and we will furthermore assess neutrophil migration in-vitro. In addition, experiments in specific transgenic MRP8-CreArp2/3-deficient mice are planned, which will provide further insights by employing the inferior vena cava flow‑restriction model that induces venous thrombosis and thereby allowing the visualization of thrombus formation and its modulation by targeted interventions.7
References
- Thienel M, Müller-Reif JB, Zhang Z, Ehreiser V, Huth J, Shchurovska K, Kilani B, Schweizer L, Geyer PE, Zwiebel M, Novotny J, Lüsebrink E, Little G, Orban M, Nicolai L, El Nemr S, Titova A, Spannagl M, Kindberg J, Evans AL, Mach O, Vogel M, Tiedt S, Ormanns S, Kessler B, Dueck A, Friebe A, Jørgensen PG, Majzoub-Altweck M, Blutke A, Polzin A, Stark K, Kääb S, Maier D, Gibbins JM, Limper U, Frobert O, Mann M, Massberg S, Petzold T. Immobility-associated thromboprotection is conserved across mammalian species from bear to human. Science. 2023; 380(6641): 178-187.
- Henson JH, Yeterian M, Weeks RM, Medrano AE, Brown BL, Geist HL, Pais MD, Oldenbourg R, Shuster CB. Arp2/3 complex inhibition radically alters lamellipodial actin architecture, suspended cell shape, and the cell spreading process. Mol Biol Cell. 2015; 26(5): 887-900.
- Glaser KM, Doon-Ralls J, Walters N, Rima XY, Rambold AS, Réategui E, Lämmermann T. Arp2/3 complex and the pentose phosphate pathway regulate late phases of neutrophil swarming. iScience. 2024; 27(1): 108656.
- Fritz-Laylin LK, Riel-Mehan M, Chen BC, Lord SJ, Goddard TD, Ferrin TE, Nicholson-Dykstra SM, Higgs H, Johnson GT, Betzig E, Mullins RD. Actin-based protrusions of migrating neutrophils are intrinsically lamellar and facilitate direction changes. Elife. 2017; 6: e26990.
- Sprenkeler EGG, Tool ATJ, Henriet SSV, van Bruggen R, Kuijpers TW. Formation of neutrophil extracellular traps requires actin cytoskeleton rearrangements. Blood. 2022; 139(21): 3166-3180.
- Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, Yavuz G, Luckner M, Ishikawa-Ankerhold H, Hennel R, Benechet A, Lorenz M, Chandraratne S, Schubert I, Helmer S, Striednig B, Stark K, Janko M, Böttcher RT, Verschoor A, Leon C, Gachet C, Gudermann T, Mederos Y Schnitzler M, Pincus Z, Iannacone M, Haas R, Wanner G, Lauber K, Sixt M, Massberg S. Migrating platelets are mechano-scavengers that collect and bundle bacteria. Cell. 2017; 171(6): 1368-1382.e1323.
- von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exl Med. 2012; 209(4): 819-835.
Impact of ICU dietary conditions on the susceptibility to hospital-acquired infections via host-microbiota and innate immune system-epithelial perturbations
Principle Investigator

Scientific interest within the context of the graduate college
Hospital-acquired infections remain one of the most pressing unresolved challenges in critical care, yet the role of nutritional support, ubiquitous in ICU management, in shaping host susceptibility through microbiota and mucosal immune perturbations has been systematically overlooked. This project offers a doctoral candidate the opportunity to investigate this clinically urgent and mechanistically underexplored question using state-of-the-art approaches spanning gnotobiotic mouse models, microbiome profiling, intestinal organoid culture, and transcriptional analyses of epithelial cells.
Project description
Hospital-acquired infections (HAI) are a major cause of death in intensive care units (ICUs). Mucosal barrier disturbances, immunosuppression and systemic lymphopenia have been identified as predisposing factors but the underlying molecular networks causing increased susceptibility to infections remain to be explored. In particular, the effect of exogenous factors, such as diet composition and formulation, on these biological processes in ICU settings has been understudied.
The microbiota has important effects on epithelial barrier function as well as on mucosal and systemic immunity which are collectively shaped by dietary input.1,2 Conventional diets contain bioactive metabolites (such as ligands for the nutrient sensors AhR, RAR, RORγt and many more) that affect the differentiation and function of immune cells.3,4 In particular, ILCs have been recently identified as a major checkpoint for immune-epithelial crosstalk and epithelial homeostasis.5 We recently demonstrated that an ILC-macrophage module is an important determinant for inflammatory output and epithelial integrity.6 We also showed that the microbiota controls steady-state type I IFN (IFN-I) levels required for metabolic fitness of myeloid cells and for effective antibacterial or antiviral immunity.7,8 Critically ill patients in ICUs often receive broad spectrum antibiotics and enteral or parenteral nutritional support that has been associated with impaired host immune defenses and susceptibility to nosocomial infections.9 Recently, we showed that feeding mice with a chemically defined parenteral diet used in ICUs abolished the mucosal germinal center reaction and IgA response when compared to a conventional diet.10 The presence of bacteria-derived components in the diet, such as lipopolysaccharide (LPS), promoted germinal center activity and dendritic cell trafficking in dietary mucosal immune responses. The effects of LPS were only reproduced when it was presented within colloidal liposomes rather than in dispersed solution. Moreover, it has been shown that exclusive enteral nutrition reduced inflammatory bowel disease (IBD) in mice by increasing the abundance of E. rectale and as consequence the production of isobutyrate.11 These results suggest that both the composition and the formulation of the diet affect the host immune system and potentially also the susceptibility to infections. We hypothesize that enteral diets used in ICU settings alter microbiota composition, epithelial barrier function, and innate immunity, increasing the risk of HAI.
Aim 1: How do feeding conditions mimicking ICU diets affect intestinal dysbiosis?(Ronchi) In animal models, we will analyze intestinal microbiota composition and function, and perform functional and transcriptional analyses of intestinal epithelial cells in animal models fed enteral diets and treated with broad-spectrum antibiotics. We will study the impact of mechanosensing triggered by the physical properties of solid vs. liquid food12,13 intestinal motility, nutrient absorption, barrier integrity, and food passage. We will reverse these changes through dietary modifications, such as adding bacterial products (e.g., LPS) or metabolites (e.g., AhR, RAR, RORγt ligands).
Aim 2: How do ICU feeding conditions alter the intestinal innate immune system (i.e., myeloid and innate lymphoid cells, ILCs) and the epithelial barrier? (Diefenbach) We will analyze the phenotype of myeloid and innate lymphoid cells (ILCs) in intestinal and respiratory mucosa, as well as in lymphoid organs including gut/lung-draining lymph nodes, spleen, and bone marrow. An important focus will be previously identified immune-epithelial cell and neuro-immune modules informed by cytokines (e.g., IL-22, IFN-I, IFN-III, IL-17) or neuronal factors (e.g., VIP, NMU), respectively. We aim to rescue observed changes via the same dietary modifications as in Aim 1.
Aim 3: How do such alterations of the microbiota-epithelial-immune metasystem of the intestines affect susceptibility to HAI? (joint aim) We will test whether the experimental conditions and resulting microbiota-epithelial-immune changes (Aims 1-2) affect susceptibility to hospital-acquired K. pneumoniae infections. We will also examine whether diets with altered biophysical properties increase resistance to infection.
Complementary expertise of the PIs. Our labs will contribute expertise in innate immunity and epithelial cell biology (Diefenbach) and microbiome research (Ronchi). The successful candidate will focus on Aim 1, taking ownership of how ICU-mimicking dietary conditions reshape intestinal microbiota composition and epithelial barrier integrity, and will be a central contributor to the project’s integrative scientific narrative across all three aims. The position sits at the interface of microbiology, mucosal immunology, and clinical nutrition, embedded in a collaborative environment bridging two complementary laboratories with expertise in microbiome research (Ronchi) and innate immunity and epithelial biology (Diefenbach). We are looking for a medically trained candidate with genuine curiosity about the biology underlying clinical phenomena and the ambition to develop as an independent scientific thinker.
References
- Hebbandi Nanjundappa R*, Ronchi F*, Wang J, Clemente-Casares X, Yamanouchi J, Sokke Umeshappa C, Yang Y, Blanco J, Bassolas-Molina H, Salas A, Khan H, Slattery RM, Wyss M, Mooser C, Macpherson AJ, Sycuro LK, Serra P, McKay DM, McCoy KD, Santamaria P. A gut microbial mimic that hijacks diabetogenic autoreactivity to suppress colitis. Cell. 2017; 171(3): 655-667.e17. *equal contribution
- Mamantopoulos M*, Ronchi F*, Van Hauwermeiren F, Vieira-Silva S, Yilmaz B, Martens L, Saeys Y, Drexler SK, Yazdi AS, Raes J, Lamkanfi M, McCoy KD, Wullaert A. Nlrp6- and ASC-dependent inflammasomes Do not shape the commensal gut microbiota composition. Immunity. 2017; 47(2): 339-348.e4. *equal contribution
- Gronke K, Hernández PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, Witkowski M, Tizian C, Amann L, Schumacher F, Glatt H, Triantafyllopoulou A, Diefenbach A. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. 2019; 566: 249-253.
- Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, Diefenbach A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011; 334(6062): 1561-1565.
- Diefenbach A, Gnafakis S, Shomrat O. Innate lymphoid cell-epithelial cell modules sustain intestinal homeostasis. Immunity. 202; 52(3): 452-463.
- Biniaris-Georgallis SI, Aschman T, Stergioula K, Schreiber F, Jafari V, Taranko A, Karmalkar T, Kasapi A, Lenac Rovis T, Jelencic V, Bejarano DA, Fabry L, Papacharalampous M, Mattiola I, Molgora M, Hou J, Hublitz KW, Heinrich F, Guerra GM, Durek P, Patone G, Lindberg EL, Maatz H, Hölsken O, Krönke G, Mortha A, Voll RE, Clarke AJ, Hauser AE, Colonna M, Thurley K, Schlitzer A, Schneider C, Stamatiades EG, Mashreghi MF, Jonjic S, Hübner N, Diefenbach A*, Kanda M, Triantafyllopoulou A*. Amplification of autoimmune organ damage by NKp46-activated ILC1s. Nature. 2024; 634(8035): 952-960. *equal contribution
- Schaupp L, Muth S, Rogell L, Kofoed-Branzk M, Melchior F, Lienenklaus S, Ganal-Vonarburg SC, Klein M, Guendel F, Hain T, Schütze K, Grundmann U, Schmitt V, Dorsch M, Spanier J, Larsen PK, Schwanz T, Jäckel S, Reinhardt C, Bopp T, Danckwardt S, Mahnke K, Heinz GA, Mashreghi MF, Durek P, Kalinke U, Kretz O, Huber TB, Weiss S, Wilhelm C, Macpherson AJ, Schild H, Diefenbach A*,1, Probst HC*. Microbiota-induced type I interferons instruct a poised basal state of dendritic cells. Cell. 2020; 181(5): 1080-1096.e19. *equal contribution, 1lead author
- Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, Diefenbach A. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity. 2012; 37(1): 171-86.
- Schlechte J, Zucoloto AZ, Yu IL, Doig CJ, Dunbar MJ, McCoy KD, McDonald B. Dysbiosis of a microbiota–immune metasystem in critical illness is associated with nosocomial infections. Nat Med. 2023; 29(4): 1017-1027.
- Mooser C, Ronchi F, Limenitakis JP, Kalbermatter C, Christensen S, de Agüero MG, Fuhrer T, Sauer U, Macpherson AJ, Ganal-Vonarburg SC. Diet-derived LPS determines intestinal IgA induction and repertoire characteristics independently of the microbiota. Immunity. 2025; 58(7): 1778-1793.e7.
- Vasconcelos Pereira G, Boudaud M, Wolter M, Alexander C, De Sciscio A, Grant ET, Trindade BC, Pudlo NA, Singh S, Campbell A, Shan M, Zhang L, Willieme S, Kim K, Denike-Duval T, Bleich A, Schmidt TM, Kennedy L, Lyssiotis CA, Chen GY, Eaton KA, Desai MS, Martens EC. Unravelling specific diet and gut microbial contributions to inflammatory bowel disease. Res Sq. 2023; 13:rs.3.rs-2518251.
- Wolfson RL, Abdelaziz A, Rankin G, Kushner S, Qi L, Mazor O, Choi S, Sharma N, Ginty DD. DRG afferents that mediate physiologic and pathologic mechanosensation from the distal colon. Cell. 2023; 186(16): 3368-3385.e18.
- Servin-Vences MR, Lam RM, Koolen A, Wang Y, Saade DN, Loud M, Kacmaz H, Frausto S, Zhang Y, Beyder A, Marshall KL, Bönnemann CG, Chesler AT, Patapoutian A. PIEZO2 in somatosensory neurons controls gastrointestinal transit. Cell. 2023; 186(16): 3386-3399.e15.
Characterization of natural killer cells harboring clonal hematopoiesis mutations in patients with cancer and cardiovascular diseases
Principal Investigator

Scientific interest within the context of the graduate college
The scientific focus of our lab within the framework of the graduate college centers on unraveling the multifaceted role of clonal hematopoiesis (CH) in disease prevention and inflammatory processes. CH is recognized as a pre-malignant condition that significantly increases the risk of hematologic malignancies. Beyond its oncogenic potential, CH has emerged as a critical risk factor for cardiovascular diseases such as stroke, myocardial infarction, and atherosclerosis, contributing to both initial and recurrent events. Moreover, CH is intricately linked to chronic inflammation, functioning both as a driver and a consequence of sustained immune dysregulation. By investigating these complex interconnections, our research aims to deepen the understanding of CH as a central node in disease pathogenesis and to identify novel strategies for early intervention and prevention.
Project description
Clonal hematopoiesis (CH), defined by the acquisition of somatic mutations in hematopoietic stem cells, occurs in 20% to 30% of individuals >60 years. CH is associated with a higher overall mortality and an approximately 10-fold risk for the development of hematologic malignancies.1 Reduced overall survival in individuals with CH is mainly caused by an increased rate of cardiovascular events.2 A causal relation was found in preclinical models, showing accelerated development of atherosclerosis driven by an altered function of the NLRP3/IL1β inflammasome of mutated monocytes/macrophages.3 These results pinpoint toward pleiotropic effects of mutated clones, not only affecting self-renewal and differentiation of hematopoietic stem cells but also inflammatory signaling of mature blood cells.
We and others have shown, that in addition to myeloid cells, natural killer (NK) cells are affected by somatic CH mutations to a comparable extent.4 This finding is somehow surprising as other lymphoid cells such as B- or T-cells only occasionally harbor CH mutations pointing to a pivotal role of NK cells within the lymphoid compartment. However, it remains poorly understand which NK subtypes are affected and whether the mutated NK cells harbor distinct KIR receptor repertoire and/or protein expression. The Damm lab has well-documented expertise in investigating CH in large-patient cohorts5 coupled with profound flow-cytometry and single-cell technologies and has established the entire wet- and dry-lab pipelines to successfully conduct such complex experiments.6,7 In close collaboration with experts in the field of NK cell biology (Prof. Romagnani, Charité), we will investigate the repartition and dynamics of CH mutations in these specialized lymphocytes of the innate immune system using readily available patient samples.
Aim 1: Defining the repartition of CH mutation mutations within the NK cell compartment. The repartition of mutated NK cells within the NK differentiation tree will be investigated for approximately 50 patients. Sample selection will be equally distributed between patients with cancer and cardiovascular diseases, also 10 healthy individuals with known CH mutations will be included. First, major NK cells subset will be sorted by flow-cytometry for the main NK cell subsets based on their expression of CD16, CD56 and additional cell surface markers.8 Thereafter, the mutation burden of patient-specific CH mutations will be quantified in the flow-sorted cell fractions by error-corrected target sequencing comprising 45 CH-associated genes, a fully established and automated workflow in our research group. CH mutations will be called using our in-house variant calling pipeline as recently published.6
Aim 2: Identification of CH and TI-CH related clinical phenotypes. Next, we will integrate the genomic results obtained in Aim 1 with advanced single-cell technologies in a subset of 10 patients. To achieve this, we will employ the Mission Bio Tapestri platform, which enables the simultaneous profiling of DNA variants and surface protein expression at single-cell resolution. This approach provides high genotyping efficiency and allows for the precise mapping of clonal architecture alongside phenotypic states across thousands of individual cells. By coupling genotype and immunophenotype data, we aim to resolve intrapatient heterogeneity, identify rare subclones, and link specific genetic alterations to functional cellular phenotypes. This integrative analysis will offer critical insights into the relationship between clonal evolution and immune cell states within each patient.
References
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014; 371(26): 2488-2498.
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017; 377(2): 111-121.
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andrés V, Hirschi KK, Martin KA, Walsh K. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017; 355(6327): 842-847.
- Arends CM, Galan-Sousa J, Hoyer K, Chan W, Jäger M, Yoshida K, Seemann R, Noerenberg D, Waldhueter N, Fleischer-Notter H, Christen F, Schmitt CA, Dörken B, Pelzer U, Sinn M, Zemojtel T, Ogawa S, Märdian S, Schreiber A, Kunitz A, Krüger U, Bullinger L, Mylonas E, Frick M, Damm F. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia. 2018; 32(9): 1908-1919.
- Arends CM, Liman TG, Strzelecka PM, Kufner A, Löwe P, Huo S, Stein CM, Piper SK, Tilgner M, Sperber PS, Dimitriou S, Heuschmann PU, Hablesreiter R, Harms C, Bullinger L, Weber JE, Endres M, Damm F. Associations of clonal hematopoiesis with recurrent vascular events and death in patients with incident ischemic stroke. Blood. 2023; 141(7): 787-799.
- Arends CM, Kopp K, Hablesreiter R, Estrada N, Christen F, Moll UM, Zeillinger R, Schmitt WD, Sehouli J, Kulbe H, Fleischmann M, Ray-Coquard I, Zeimet A, Raspagliesi F, Zamagni C, Vergote I, Lorusso D, Concin N, Bullinger L, Braicu EI, Damm F. Dynamics of clonal hematopoiesis under DNA-damaging treatment in patients with ovarian cancer. Leukemia. 2024; 38(6): 1378-1389.
- Wiegand L, Silva P, Noerenberg D, Christen F, Kopp K, Locher BN, Löwe P, Tilgner M, Altwasser R, Storzer V, Stein CM, Briest F, Arends CM, Frick M, Ihlow J, Dolnik A, Ishaque N, Keller U, Na IK, Penter L, Bullinger L, Hablesreiter R, Damm F. Clonal hematopoiesis and lymphoma-associated mutations in hematopoietic progenitors in B-cell non-Hodgkin lymphoma. Blood. 2026; 147(15): 1723-1734.
- Rebuffet L, Melsen JE, Escalière B, Basurto-Lozada D, Bhandoola A, Björkström NK, Bryceson YT, Castriconi R, Cichocki F, Colonna M, Davis DM, Diefenbach A, Ding Y, Haniffa M, Horowitz A, Lanier LL, Malmberg KJ, Miller JS, Moretta L, Narni-Mancinelli E, O’Neill LAJ, Romagnani C, Ryan DG, Sivori S, Sun D, Vagne C, Vivier E. High-dimensional single-cell analysis of human natural killer cell heterogeneity. Nat Immunol. 2024;25(8): 1474-1488.
Expression pattern and function of IL-12 receptor in the central nervous system in neurodegeneration and aging
Prinicipal Investigator

PD Dr. Marina Jendrach
Scientific interest within the context of the graduate college
Our project directly aligns with the central mission of the Else Kröner-Fresenius Graduate College Re-Thinking Health, which aims to establish a novel, mechanistic understanding of health as an active, regulated biological state and to identify molecular targets for prevention. Rather than focusing solely on disease mechanisms, the project investigates how immune-mediated signaling pathways contribute to the maintenance of neuronal integrity and functional resilience in the central nervous system. Specifically, this project addresses the role of Interleukin-12 (IL-12) signaling in neurons and glial cells in the context of aging and neurodegeneration. By dissecting the cell type-specific functions of the IL-12 receptor, particularly in Parvalbumin-positive interneurons, the study explores whether immune-derived signals contribute to adaptive, health-preserving processes in the brain. This approach reflects the Graduate College’s conceptual framework that health is maintained by distinct molecular pathways, including those mediating adaptation to environmental and internal stressors.
Importantly, the project integrates both human and murine data to identify molecular signatures associated with resilient versus maladaptive neuroinflammatory responses. In doing so, it contributes to defining early deviations from a healthy state, a key objective of the program. Furthermore, by identifying cell-specific signaling mechanisms that modulate neuronal survival, network stability, and myelination, the project may uncover novel molecular targets for preventive strategies in age-associated neurodegenerative diseases such as Alzheimer’s disease. Overall, this work exemplifies the transition from a disease-centered to a health-centered research paradigm by investigating how immune-neuronal interactions shape brain health, resilience, and the prevention of neurodegenerative pathology.
Project description
Microglia, the resident myeloid cells of the central nervous system (CNS), undergo pronounced phenotypic changes during aging and in neurodegenerative disorders. While early microglial activation can exert neuroprotective effects, sustained activation promotes chronic neuroinflammation through the release of neurotoxic mediators, including pro-inflammatory cytokines.1,2 Interleukin-12 (IL-12) is one of these proinflammatory cytokines, produced primarily by activated microglia in the CNS. Elevated IL-12 levels have been reported in the brains of patients with Alzheimer’s disease (AD) as well as in AD-like mouse models.3,4
In acute neuroinflammatory conditions characterized by strong cytokine responses, such as multiple sclerosis, IL-12 has been shown to exert neuroprotective effects on neurons, though not on oligodendrocytes.5 In contrast, during chronic neuroinflammation, as observed in AD mouse models and likely also in aging, IL-12 deficiency results in a marked reduction of AD-like pathology, including decreased amyloid-β deposition and altered neuroinflammation.6,7 Notably, IL-12 deficiency confers neuroprotection in this context, particularly for Parvalbumin (PV)-positive GABAergic interneurons, a neuronal population that is especially vulnerable in early Aβ pathology.4,8
Preliminary data from our group indicate a region-specific expression pattern of the IL-12 receptor in neurons (Heppner lab, unpublished). Based on this, the first aim of the project is to systematically assess IL-12 receptor expression in murine and human brain tissue across aging and disease conditions. However, as IL-12 receptors are expressed not only on neurons but also on oligodendrocytes, conventional IL-12 knockout models do not allow discrimination between cell type-specific effects. Given that oligodendrocytes are responsible for myelination and that neuronal activity can influence this process,9 the observed neuroprotective phenotype in IL-12-deficient models may, at least in part, result from secondary effects on myelination. For example, IL-12-stimulated neurons can release trophic factors that support oligodendrocyte function.5 To address this limitation, the second aim is to selectively abrogate IL-12 signaling in PV-positive neurons by deleting the IL-12 receptor using a PV-Cre mouse model (constitutive deletion). This approach will enable a cell type-specific and temporally resolved analysis of IL-12 signaling in this vulnerable neuronal population during aging and in AD-like pathology.
Aim 1: Translational correlation of IL12 receptor expression. Models: IL12rb1 and rb2 expression in various existing mouse models and age groups and in human samples from the Charité Brain Bank; Methods: RNAscope; immunohistochemistry; confocal microscopy, image analysis
Aim 2: Effects of the loss of IL-12 signaling in PV interneurons in aging and AD-like mice. Models: PV-Cre.IL12rb2 and APPPS1.PV-Cre.IL12rb2 mice. Mouse colonies are established and animal permit is granted; Methods: Analysis of neuroinflammation, neuronal changes and Alzheimer’s pathology: protein extraction, Mesoscale/ELISA, immunohistochemistry, Western blot, myelinization, (electrophysiology in cooperation with the Geiger Lab).
References
- Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015; 16(6): 358-372.
- Cai Y, Liu J, Wang B, Sun M, Yang H. Microglia in the neuroinflammatory pathogenesis of Alzheimer’s disease and related therapeutic targets. Front Immunol.2022; 13: 856376.
- Bruno M, Bonomi CG, Castelli A, Izzi F, Placidi F, Falletti S, Mercuri NB, Motta C, Martorana A. Cerebrospinal fluid cytokine levels affect electroencephalographic activity in Alzheimer’s disease. J Alzheimers Dis Rep. 2025; 9: 25424823241306772.
- Schneeberger S, Kim SJ, Geesdorf MN, Friebel E, Eede P, Jendrach M, Boltengagen A, Braeuning C, Ruhwedel T, Hülsmeier AJ, Gimber N, Foerster M, Obst J, Andreadou M, Mundt S, Schmoranzer J, Prokop S, Kessler W, Kuhlmann T, Möbius W, Nave KA, Hornemann T, Becher B, Edgar JM, Karaiskos N, Kocks C, Rajewsky N, Heppner FL. Interleukin-12 signaling drives Alzheimer’s disease pathology through disrupting neuronal and oligodendrocyte homeostasis. Nat Aging. 2025; 5(4): 622-641.
- Andreadou M, Ingelfinger F, De Feo D, Cramer TLM, Tuzlak S, Friebel E, Schreiner B, Eede P, Schneeberger S, Geesdorf M, Ridder F, Welsh CA, Power L, Kirschenbaum D, Tyagarajan SK, Greter M, Heppner FL, Mundt S, Becher B. IL-12 sensing in neurons induces neuroprotective CNS tissue adaptation and attenuates neuroinflammation in mice. Nat Neurosci. 2023; 26(10): 1701-1712.
- Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat Med. 2012; 18(12): 1812-1819.
- Eede P, Obst J, Benke E, Yvon-Durocher G, Richard BC, Gimber N, Schmoranzer J, Böddrich A, Wanker EE, Prokop S, Heppner FL. Interleukin-12/23 deficiency differentially affects pathology in male and female Alzheimer’s disease-like mice. EMBO Rep. 2020; 21(3): e48530.
- Chen YB, Jiang B, Zhuang K, Wang H, Peng HM, Zhong BW, Xie WT, Chen KW, Zou TT, Wang Y, Yang HL, Yang Q, Zhou J, Zhong L, Zhang LH, Zhang J. Hyperactivity of subicular parvalbumin interneurons drives early amyloid pathology and cognitive deficits in Alzheimer’s disease. Mol Psychiatry. 2025; 30(12): 5777-5789.
- Zhou Y, Zhang J. Neuronal activity and remyelination: new insights into the molecular mechanisms and therapeutic advancements. Front Cell Dev Biol. 2023; 11: 1221890.
Tissue niches and signals regulating lymphocyte maintenance
Principle Investigator

Dr. Christina Stehle
Scientific interest within the context of the graduate college
The innate immune system develops early in parallel with tissue formation, suggesting its role extends beyond host defense. In particular, tissue-resident innate lymphoid cells (ILCs) colonize tissues during fetal life and acquire effector functions prenatally acting as early-life sensors of the tissue environment. Our research focuses on understanding how the immune system, and ILCs in particular, contribute to the active maintenance of tissue homeostasis and how tissue-derived signals shape ILC differentiation, maintenance, and function. By defining these pathways, we aim to understand the molecular transition from a state of health to the earliest stages of pathology.
Project description
Lymphocytes comprise both adaptive and innate populations that arise from hematopoietic precursors. While these precursors originate in the fetal liver during embryogenesis and in the bone marrow after birth, accumulating evidence from our previous studies in mice and humans indicates that peripheral tissues function as active sites of lymphopoiesis, with local stromal, endothelial and epithelial cells providing nonredundant signals that support lymphocyte differentiation, maintenance, and function in situ.
In this context, the common y-chain cytokine IL-7 plays a central role in the survival, differentiation, and activation of both adaptive and innate lymphocyte populations. IL-7 signaling has established as key regulator of ILC homeostasis and IL-22 production, both of which are essential for maintaining the barrier integrity of the intestine. Using a combination of single-cell sequencing, in vitro systems, and in vivo analyses in established genetic mouse models, this project aims to define the cellular niches and molecular signals that sustain tissue-resident lymphocytes. we seek to determine how disruption of these circuits contributes to loss of tissue homeostasis and the development of immune-mediated pathology. To answer these questions, the project will have the following aims:
Aim 1: Define the intestinal ILC niche. To provide insights into the gene expression profiles and spatial organization of cells that facilitate health-maintaining interactions, we will characterize the IL-7-ILC niche during homeostatic balance. We aim to analyse unbiased single cell transcriptomic datasets of intestinal cell types (epithelial, endothelial, stromal cells, and ILCs) at steady-state and following environmental changes influenced by nutrient fluctuations along the proximal-distal axis of the small intestine. The goal is to identify the local cellular sources of IL-7 creating the anatomical niche required for the maintenance of intestinal ILCs. Methodological approaches will include bioinformatic analyses (R-Studio/Seurat), qPCR validations, and flow cytometry.
Aim 2: Elucidate the role of intestinal IL-7 sources for ILC composition. In our preliminary data, we have generated a conditional deletion of IL-7 selectively in the intestinal epithelium (VilCre x Il7fl/fl mice), where we observed a selective reduction of type 3 ILCs in the intestinal lamina propria, suggesting a non-redundant role for epithelial-derived IL-7 in maintaining tissue equilibrium. To gain a comprehensive understanding of the spatial requirements of IL-7, we will comparatively analyze lymphocyte subsets in mice that selectively ablate other sources of IL-7 in stromal (Pdgfra-cre) and endothelial (Cdh5-cre) compartments. All mouse lines are already established.
Overall, this project addresses the fundamental question of how tissue-resident immune cells act as actors of homeostasis. By comprehensively characterizing the small intestinal ILC niche, we aim to define the molecular requirements for “health” and identify the pathways that prevent the transition towards chronic inflammatory disease.
References
- Juelke J, Romagnani C. Differentiation of human innate lymphoid cells (ILCs). Curr Opin Immunol. 2016; 38: 75-85.
- Montaldo E, Teixeira-Alves LG, Glatzer T, Durek P, Stervbo U, Hamann W, Babic M, Paclik D, Stölzel K, Gröne J, Lozza L, Juelke K, Matzmohr N, Loiacono F, Petronelli F, Huntington ND, Moretta L, Mingari MC, Romagnani C. Human RORγt+CD34+ cells are lineage-specified progenitors of group 3 RORγt+ innate lymphoid cells. Immunity. 2014; 41(6): 988-1000.
- Stehle C, Rückert T, Fiancette R, Gajdasik DW, Willis C, Ulbricht C, Durek P, Mashreghi MF, Finke D, Hauser AE, Withers DR, Chang HD, Zimmermann J, Romagnani C. T-bet and RORα control lymph node formation by regulating embryonic innate lymphoid cell differentiation. Nat Immunol. 2021; 22(10): 1231-1244.